
As an undergraduate student excited about AI for healthcare applications, I was thrilled to be joining the NVIDIA Clara Deploy team for an internship. It was the perfect combination: the opportunity to work at a leading technology company enabling the acceleration and adoption of AI while contributing to a team building the future (and the present!) of AI deployment for healthcare. The next few months were filled with learning from brilliant yet humble colleagues, picking up new skills like CUDA programming, and the opportunity to focus on unique technical challenges posed by histopathology data.
What is Clara Deploy?
The Clara Deploy SDK is a container-based, cloud-native development and deployment framework for multi-AI and multidomain workflows in smart hospitals. It enables you to define container-based pipelines consisting of multiple stages, each stage defined by an operator. A pipeline consists of multiple operators and is a directed acyclic graph (DAG) from the data source to the data sink. Each operator is a step of the pipeline, such as loading input, preprocessing, AI inference, and so on.
As I explored setting up the NVIDIA Clara Deploy platform and running AI inference pipelines, I gained firsthand experience in the challenges of deploying AI workflows, particularly in standardizing workflows and scaling up execution. While running digital pathology pipelines, I gained awareness of the performance bottleneck of I/O and preprocessing steps that are usually not GPU-accelerated. This influenced my choice to focus on accelerating preprocessing filters for digital pathology during my internship.
What is cuCIM?
cuCIM is a RAPIDS library for accelerated n-dimensional image processing and image I/O, with a focus on medical imaging applications. cuCIM consists of I/O, file system, and operation modules. Operations in cuCIM can be extended using a plug-in architecture. cuCIM is uniquely positioned to be a leading library for medical image-processing applications, and I am excited to have gained exposure to and contributed to it during my time at NVIDIA.
Project motivation
A significant challenge in the digitization of histopathology analysis is the stain variation observed in pathology images. These images can have large variations in staining caused by multiple factors, including stain vendors, storage conditions, staining protocols, digital scanners, and so on.
Given the range of factors, it is impractical to control for staining variation during image acquisition. Instead, an image preprocessing step called stain normalization is often used to algorithmically standardize image staining. A stain normalization filter accepts as input a source image and a target image. The source image is to be stain normalized, and the target image contains the ideal stain, to be transferred to the source image. Ultimately, a normalized source image is returned as output.
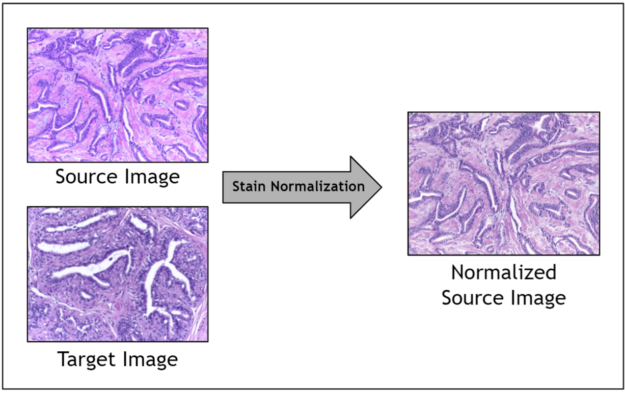
Prior work has shown that stain normalization used as a preprocessing step in digital pathology AI pipelines can shorten training time, improve accuracy, and enable data from different sources to be used together. Because you are operating in a relatively small data regime due to the scarcity of stained pathology images, stain normalization enables you to optimize the signal obtained amidst noisy stain variations.
However, prior implementations of stain normalization were relatively slow as they were not GPU-accelerated. There was an opportunity to implement a GPU-accelerated stain normalization algorithm and enable fast and effective preprocessing for digital pathology AI pipelines.
Accelerating stain normalization for digital pathology
Stain normalization methods fall into three broad categories:
- Global color normalization
- Color normalization after stain deconvolution
- Color transfer using deep networks
For more information, see Stain Color Adaptive Normalization (SCAN) algorithm: Separation and standardization of histological stains in digital pathology.
I chose to focus on stain deconvolution-based methods, as prior literature showed greater performance compared to global color normalization and better theoretical guarantees regarding the maintenance of biological structure integrity compared to deep network-based methods.
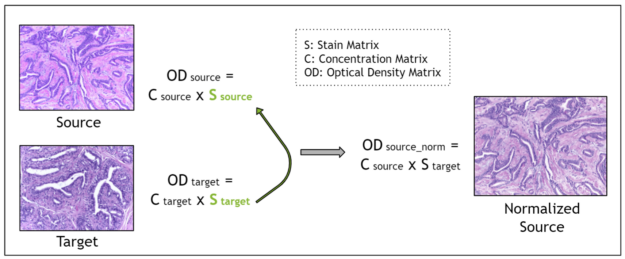
Stain deconvolution-based methods assume that each image is characterized by a stain matrix, which contains the red, green, blue (RGB) values for each of the two stains in H&E stained images: hematoxylin and eosin.
Using the Beer-Lambert law, an RGB image is transformed into an optical density image. Then, the optical density image may be related to the product of a pixel concentration matrix and the stain matrix for that image. The pixel concentration matrix indicates the concentration of each stain for each pixel. If the stain matrix is estimated, done here with the Macenko method, then the concentration matrix may be obtained.
Finally, for stain normalization, the stain matrix of a source image is replaced with the stain matrix of a target image. This serves the purpose of transferring the stain profile from the target image to the source image. Because the concentration matrix of the source image is unchanged, the morphology of the biological structures is maintained. The Macenko method for estimating the stain matrix is an unsupervised method using the singular value decomposition.
I designed and implemented a filter for the Macenko method for stain normalization in CuPy, after modifying an existing version in NumPy. Next, I compared the performance of the two.
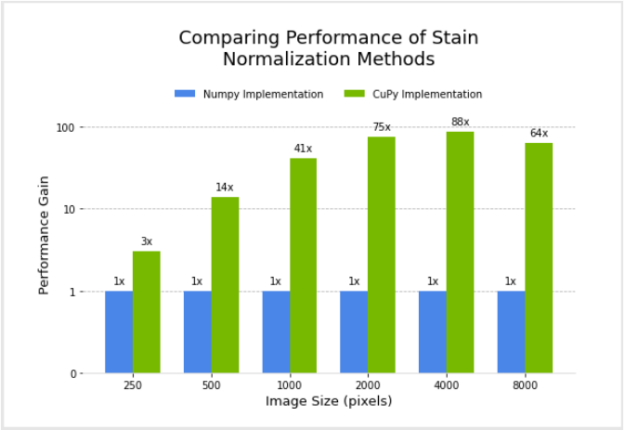
Figure 3 shows the relative performance of the NumPy and CuPy implementations of stain normalization for different image sizes, using an NVIDIA DGX-1. Performance for the CuPy implementation is plotted in terms of acceleration factor relative to the NumPy implementation.
Given the goal of enabling GPU-accelerated stain normalization to be used as a preprocessing step for digital pathology pipelines, I began the integration of this filter as a transform (array-based and dictionary-based) into MONAI. MONAI is an open-source, PyTorch-based framework for deep learning in medical imaging. After being fully integrated, the stain normalization transform can be added to pathology pipelines in Clara Train or MONAI.
Acceleration of color conversion filter
Next, I worked on implementing the color conversion rgb2hed function in CUDA C++, which is a commonly used function available in scikit-image and the cuCIM Python layer, among other libraries. Color space conversion from RGB to HED is closely related to stain normalization, as this function involves obtaining stain concentration values, assuming that the stain vectors are a constant, precalculated approximation. This ignores variations between the staining of different images. This function is to be integrated into cuCIM through a C++ based operator plugin mechanism.
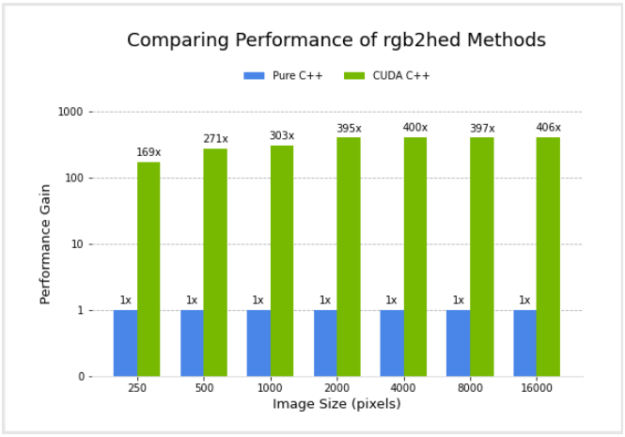
I compared the performance of a pure C++ implementation and the CUDA C++ implementation. Figure 4 shows the relative performance of the two versions, for different image sizes, using an NVIDIA GV100 GPU and Intel(R) Core(TM) i7-7800X CPU. Performance for the CUDA C++ implementation is plotted in terms of acceleration factor relative to the pure C++ implementation.
It’s important to note that the performance gains do not account for any transfer of data to and from the GPU. I did this because I am considering the common scenario where data transfers are minimized by remaining on the GPU for several subsequent operations in an image processing workflow, with transfer back to the host occurring only at the end.
Conclusion
In summary, my internship project was focused on accelerating color conversion filters for digital pathology. Specifically, I worked on designing and implementing the Macenko stain normalization method, using CuPy for GPU-acceleration. I began the integration of this into MONAI as a transform, for future use as a preprocessing step for digital pathology pipelines. Next, I worked on implementing the color conversion rgb2hed function in CUDA C++, to be integrated into cuCIM through a C++ based operator plugin mechanism.
Both the CuPy implementation of Macenko stain normalization and the CUDA C++ implementation of the rgb2hed function showed significant performance gains compared to the NumPy version and pure C++ version, respectively. The stain normalization preprocessing time for training a pipeline over 500 epochs with a dataset of 250 images and image size of 4000 by 4000 pixels is roughly estimated at 13 days with the NumPy-based filter. It decreases to 3.5 hours for the CuPy-based filter.
Ultimately, accelerating pre– and post-processing filters for digital pathology can improve the performance of deep learning pipelines in digital pathology, expedite the adoption of digital pathology, and enable AI to revolutionize pathology.
