GROMACS, a scientific software package widely used for simulating biomolecular systems, plays a crucial role in comprehending important biological processes important for disease prevention and treatment. GROMACS can use multiple GPUs in parallel to run each simulation as quickly as possible.
Over the past several years, NVIDIA and the core GROMACS developers have collaborated on a series of multi-GPU and multi-node optimizations.
In this post, we showcase the latest of these improvements, made possible through the enablement of GPU Particle-mesh Ewald (PME) decomposition with GPU direct communication: a feature available in the new GROMACS 2023 release version. We observe up to 21x performance improvements enabled through this work.
Implementation of improved multi-node performance
In a previous post, we presented optimizations to multi-GPU scalability within a single node, including the development of GPU direct communications. We described how GROMACS typically assigns one GPU to PME long-range force calculations (performed through transformations to Fourier space), with the remaining GPUs used for short-range particle-particle (PP) force calculations (performed directly in real space).
We also described developments to perform communications directly between these GPUs: halo-exchanges between the multiple PP GPUs plus PP-PME communications. We described this work in more detail in the paper, Heterogeneous parallelization and acceleration of molecular dynamics simulations in GROMACS.
GPU direct communications with MPI
In the GROMACS 2022 release version, the GPU-direct communication feature was extended to support compatibility with CUDA-aware MPI, to enable GPU-direct communications across multiple nodes (as well as within each node). For more information about how to activate this feature, see the How to run section later in this post.
However, there still existed a scalability limitation associated with the restriction to a single GPU for PME. While it was possible to add more GPUs to tackle the PP force calculations, it wasn’t possible to scale most simulations beyond a few nodes. When scaling up, the single PME GPU becomes the limiting factor at some point. The PME calculation must be decomposed across multiple GPUs to enable further scaling.
PME GPU decomposition
This single PME GPU limitation has been lifted in the brand new GROMACS 2023 release version through the introduction of PME GPU decomposition. This leverages the new NVIDIA cuFFTMp library, a library able to perform the required fast Fourier transforms (FFTs) in a distributed way across multiple GPUs within and across compute nodes.
cuFFTMp, in turn, uses NVSHMEM, a parallel programming interface enabling fast one-sided communications. It can make efficient use of intra– and inter-node interconnects to perform the all-to-all communications required by distributed 3D FFTs.
By integrating this new functionality, GROMACS can now compute multiple PME ranks in the simulation, providing dramatically enhanced scalability and performance. For portability, the implementation also supports heFFTe, an alternative to cuFFTMp. We find cuFFTMp to be up to 2x faster and so we focus on that for the remainder of this post.
This work involved several complex adaptations to the way that PME communications are handled in GROMACS, and enablements to interface with the new cuFFTMp functionality.
Several algorithmic improvements were performed to facilitate this new functionality. This includes the introduction of pipelining to enhance the overlap of computation and communication, and a parallel implementation redesign to use a grid overlap reduction mechanism instead of the redistribution of data before PME computation.
For more information, see the GROMACS GitLab issues Support PME decomposition in CUDA backend and Optimize CUDA PME Decomposition.
Performance results
In this section, we showcase performance results using the new performance features. We used the following test cases:
- Satellite Tobacco Mosaic Virus (STMV) has around 1M atoms and is described in Heterogeneous parallelization and acceleration of molecular dynamics simulations in GROMACS. It is available from Supplementary Information for Heterogeneous Parallelization and Acceleration of Molecular Dynamics Simulations in GROMACS.
- BenchPEP-h has around 12M atoms and is available from the Max Planck GROMACS Benchmark Suite.
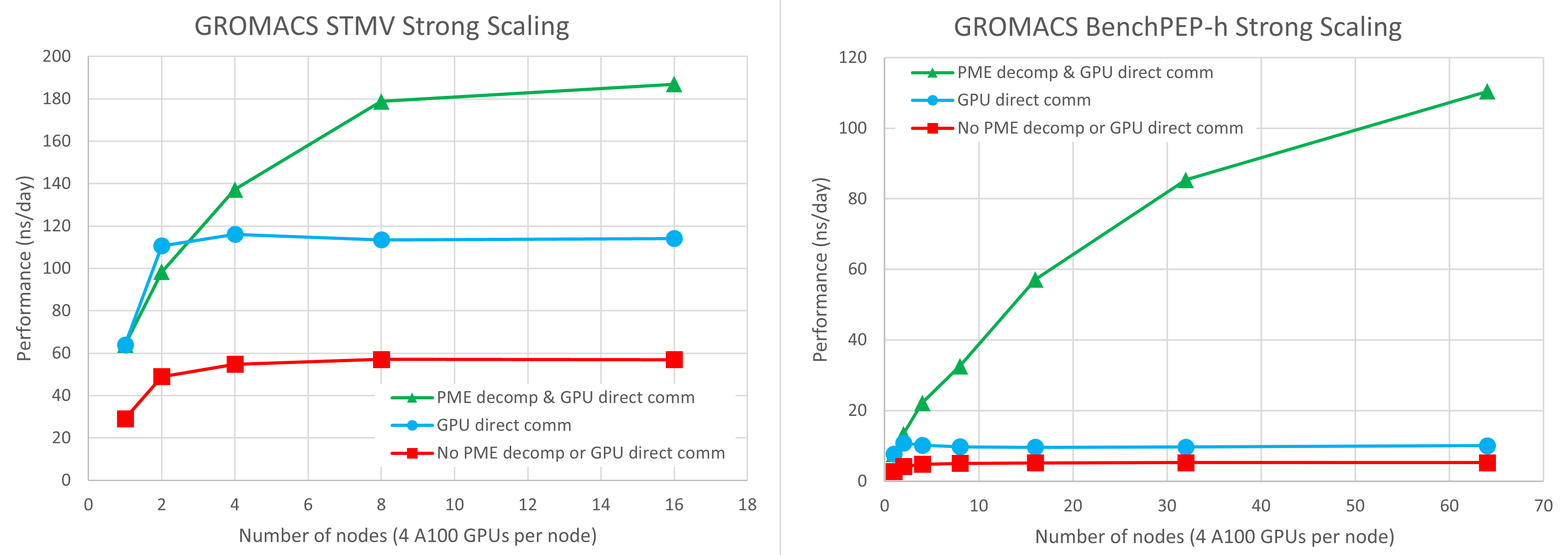
In Figure 1, we used four NVIDIA A100 GPUs per node on the Selene DGX-A100 cluster. For more information, see the next section in this post. Red squares, blue circles, and green triangles denote results using the legacy code path, GPU-direct communication only, and GPU-direct communication combined with GPU PME decomposition, respectively.
You can see that the introduction of GPU direct communications gives a 2-3x speedup over the legacy code path where communications are staged through CPU memory. With GPU direct communications, the benchmarks can scale to around two nodes, but at that point, the single PME GPU becomes the limiting factor and no further scaling is possible.
The results combining GPU direct communication with PME GPU decomposition use one PME GPU per node, which we find to give the best balance. The remaining three GPUs on each node are dedicated to the PP short-range force calculations.
On a single node, there is no effect from PME GPU decomposition, as you still only have one total PME GPU.
On two nodes, the use of two PME GPUs gives performance similar to that using a single PME GPU. The former is slightly slower for STMV and slightly faster for benchPEP-h.
At this scale, the critical path is still sensitive to the PP workload, so experimentation is necessary to find the best-performing configuration. For example, we sacrificed a PP GPU to enable the extra PME GPU. However, as you further scale up the number of nodes, the critical path is purely determined by PME performance, and the PME GPU decomposition enables much better scalability.
The STMV case scales well up to eight nodes, at which scale the results are 3x faster than the legacy code path, and 1.6x faster than the results without GPU PME decomposition.
On 16 nodes, for STMV, you see a slight improvement over eight nodes, but with much less parallel efficiency. The larger benchPEP-h case scales well up to the largest 64-node (256-GPU) configuration tested, where it is 21x faster than the legacy code path, and 11x faster than the results without GPU PME decomposition.
How to build and run GROMACS
In this section, we provide details on how we ran GROMACS with PME GPU decomposition to obtain the performance results.
We installed NVIDIA HPC SDK 22.11 by following the download and installation instructions on the website. With GROMACS 2023, we do not recommend any later version of the HPC SDK due to compatibility issues that will be addressed with future developments.
Obtain the GROMACS 2023 release version as follows:
git clone https://gitlab.com/gromacs/gromacs.git cd gromacs git checkout v2023
Build GROMACS as follows:
# make a new directory to build in
mkdir build
cd build
# set the location of the math_libs directory in the NVIDIA HPC installation
HPCSDK_LIBDIR=/lustre/fsw/devtech/hpc-devtech/alang/packages/nvhpc/nvhpc_2022_2211_Linux_x86_64_cuda_11.8-install/Linux_x86_64/2022/math_libs
# build the code with PME GPU decomposition with cuFFTMp enabled,
# in an environment with a CUDA-aware OpenMPI installation
# (see https://manual.gromacs.org/current/install-guide/index.html)
cmake \
../ \
-DGMX_OPENMP=ON -DGMX_MPI=ON -DGMX_BUILD_OWN_FFTW=ON \
-DGMX_GPU=CUDA -DCMAKE_BUILD_TYPE=Release -DGMX_DOUBLE=off \
-DGMX_USE_CUFFTMP=ON -DcuFFTMp_ROOT=$HPCSDK_LIBDIR
make -j 8 #Build using 8 CPU threads. Can increase this if you have more CPU cores available.
The HPCSDK_LIBDIR variable is set to the Linux_x86_64/2022/math_libs subdirectory of the HPC SDK installation.
Obtain the input file as follows:
wget https://zenodo.org/record/3893789/files/GROMACS_heterogeneous_parallelization_benchmark_info_and_systems_JCP.tar.gz tar zxvf GROMACS_heterogeneous_parallelization_benchmark_info_and_systems_JCP.tar.gz ln -s GROMACS_heterogeneous_parallelization_benchmark_info_and_systems_JCP/stmv/topol.tpr .
We benchmarked on the NVIDIA Selene cluster, which consists of multiple DGX-A100 servers, each with eight A100-SXM4 GPUs, connected through Infiniband. However, in line with more typical HPC installations, we only used four GPUs per node.
We ran the code after copying the STMV topol.tpr input file to the working directory, using the standard job scheduling method (Slurm in this case). We provide the full submission script used on Selene, which includes detailed commentary.
To use PME GPU decomposition, set the following variables:
# Specify that GPU direct communication should be used export GMX_ENABLE_DIRECT_GPU_COMM=1 # Specify that GPU PME decomposition should be used export GMX_GPU_PME_DECOMPOSITION=1
Specify the total number of PME GPUs through the -npme <N> flag to mdrun. Without PME GPU decomposition, N is 1 as you can only use a single PME GPU.
With decomposition, you set N to the number of nodes in use, to specify one PME GPU per node, with the other three GPUs in each node dedicated to PP. This division typically gives a good balance given the relative computational expense of the PP and PME workloads, but experimentation is recommended for any specific case.
Summary
In this post, we described our work in enhancing multi-node scalability of GROMACS, we demonstrated the performance improvements of up to 21x, and we detailed how to use these new features. This enables researchers to continue to push the boundaries in their knowledge of crucial biological processes.
We continue to improve GROMACS through a range of developments, targeting not only further enhanced scalability but also the performance and efficiency within each compute node.
GROMACS 2023 features another exciting development where cutting-edge CUDA Graphs technology has been exploited to enhance performance, particularly for small scientific systems.
Try scaling your own GROMACS case to many GPUs by following these instructions and accelerate your scientific research.
Where can you learn more? Join the GROMACS forum.