As the world battles to reach a scientific breakthrough in the fight against COVID-19, scientists are turning to computing resources to accelerate their research. To help make the process for scientists more accessible, we’re spotlighting a few of the GPU-accelerated applications that developers can use right now in the fight against this virus.
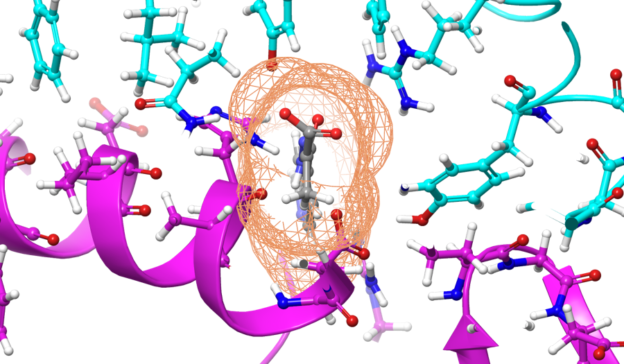
Applications like AMBER, GROMACS, NAMD, and LAMMPS are some of the popular molecular dynamics simulation applications that leverage Newton’s laws to evaluate molecular motion at the atomic level.
Today, these applications are more pivotal than ever in the fight against COVID-19. Molecular dynamics simulations offer accurate approximations of real molecular behavior, which is useful in different stages of drug development. They are used to simulate proteins and lipids and they shed light on how drugs might bind to a protein.
These molecular modelling applications are well-suited for GPUs because they lend themselves to data-parallel implementations. Simulations in these applications can run for several hundred nanoseconds. The ability to run longer simulations in less time is of extreme importance because it can show that the drug was able to modify the behavior of the protein without causing any side effects. For example, events such as folding and docking can take micro- and milliseconds to occur. A simulation that gets cut short because of limited compute power may miss these important aspects of protein and compound simulations.
GROMACS—one of the most widely used HPC applications—has received a major upgrade with the release of GROMACS 2020. The new version includes exciting new performance improvements (up to 3x faster) resulting from a long-term collaboration between NVIDIA and the core GROMACS developers.
To learn more about running the latest GPU-optimized version of GROMACS, see Creating Faster Molecular Dynamics Simulations with GROMACS 2020, published recently on the NVIDIA Technical Blog.
The NVIDIA HPC Application Performance page provides performance details by datasets and system configurations that developers can refer to as a starting point.
Scientific community battles to disable COVID-19
When the virus first broke out, researchers started using the world’s fastest computing resources to run their simulations.
Using molecular dynamics applications like GROMACS on the world’s fastest supercomputer, Summit, which is powered by more than 27,000 NVIDIA V100 GPUs, Oak Ridge National Laboratory (ORNL) researchers identified 77 small-molecule drug compounds.
Those compounds are likely to bind to the glycosylated spike (S) protein, which is how the virus enters host cells.
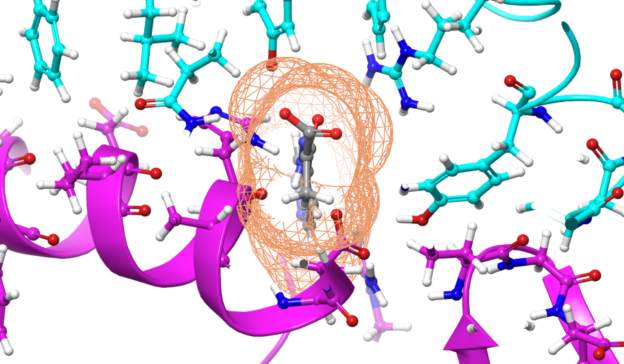
The good news is that the same NVIDIA V100 GPUs used in that project are available on the cloud to run GROMACS and the many other GPU-accelerated applications that can be used to explore solutions from multiple directions.
Helping researchers accelerate their work on the cloud
Democratizing access to supercomputing power in the science community has been the NVIDIA vision with the CUDA ecosystem. The NGC registry of containers provides easy access and simplifies deployments of scientific applications on GPU-powered systems, including cloud instances.
Several cloud service providers (CSP) have pledged support to find a cure for COVID-19. The ability to run applications like GROMACS, NAMD, and LAMMPS on NVIDIA V100 or T4 GPUs in those services can dramatically jumpstart the time for scientific computing.
Join the fight against COVID-19
Take advantage of the various molecular dynamics and genomics applications, healthcare SDK, and machine learning software from NGC to ramp up your research and fight COVID-19 on NVIDIA V100 and T4-powered cloud instances from your CSP.
Related NVIDIA corporate blog post: NVIDIA Brings GPU, HPC and AI Expertise to COVID-19 Battle
