
Many organizations are using NVIDIA Clara Parabricks for fast human genome and exome analysis for large population projects, critically ill patients, clinical workflows, and cancer genomics projects. Their work aims to accurately and quickly identify disease-causing variants, keeping pace with accelerated next-generation sequencing as well as accelerated genomic analyses.
Most recently, two peer-reviewed scientific publications in August and September highlight the speed, accuracy, and cost savings of Clara Parabricks for de novo and pathogen workflows.
Genome variant identification to track Malaria transmission
Lead Purdue University researcher Dr. Giovanna Carpi and her team sought to understand the performance of Clara Parabricks relative to existing methods used by the Malaria community for variant identification to track malaria transmission and monitor antimalarial drug resistance using 1,000 malaria genomes.
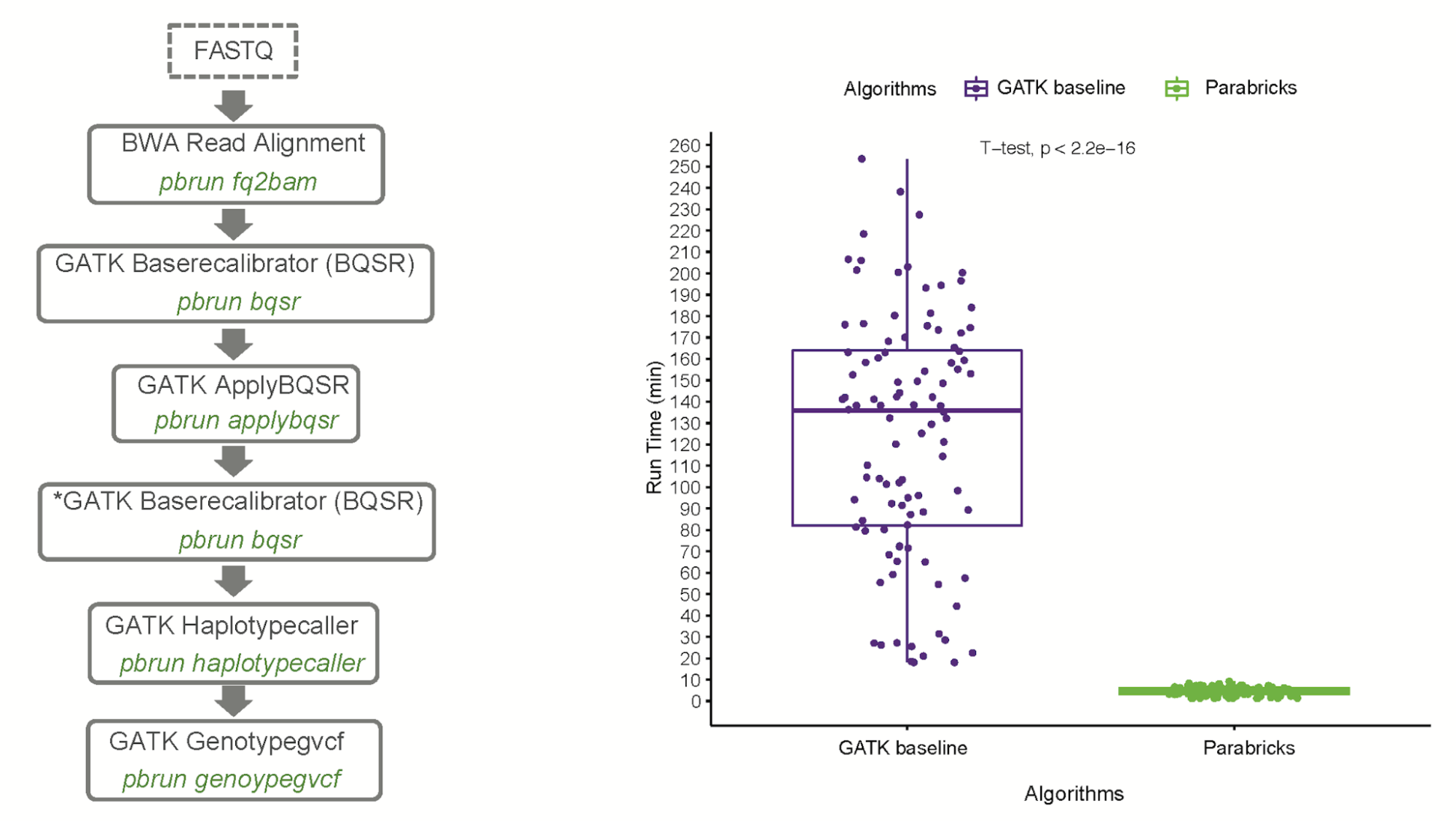
Dr. Carpi, who has been researching pathogen genomics for many years, demonstrated a 27x increase in analysis speed and a 5x decrease in cost compared to the CPU conventional pipeline, while delivering 99.9% accuracy. The malaria genome is relatively large (24 MB) and AT-rich, which makes it quite challenging to analyze. Dr. Carpi used publicly available data from the MalariaGEN consortium, which were raw reads from Illumina. The research is presented in A GPU-Accelerated Compute Framework for Pathogen Genomic Variant Identification to Aid Genomic Epidemiology of Infectious Disease: A Malaria Case Study, published in Briefings in Bioinformatics.
The ability to sequence and analyze whole-genome pathogens quickly helps public health officials understand the spread of a disease, drug resistance, and also new variants’ transmissibility and severity. The World Health Organization (WHO) reported 241 million cases of malaria in 2020 compared to 227 million cases in 2019, and an estimated 627,000 deaths in 2020—an increase of 69,000 deaths over the previous year.
Malaria is caused by Plasmodium parasites that are transmitted to people through the bites of infected female Anopheles mosquitoes. Africa carries a disproportionately high share of the global malaria burden, with children under five years of age accounting for 80% of the total deaths in the region.
Dr. Carpi noted, “The ability to generate analysis-ready variant outputs in less than five minutes with greater than 99.9% accuracy for large-scale whole-genome Plasmodium studies at lower costs, remarkably reduces the computational bottleneck that most malaria genomics programs currently face, and facilitates decentralized bioinformatics analyses in endemic countries.” Visit malaria-parabricks-pipeline on GitHub to download this Clara Parabricks workflow for malaria and to learn more.
Discovering de novo variants in autism patients
Separately, Dr. Tychele Turner and her team from Washington University in St. Louis developed a fast genomics workflow for discovering de novo variants (DNVs) in autism patients using GPU-accelerated Clara Parabricks. Dr. Turner is a geneticist/genomicist with a deep interest in understanding the genetic architecture of human disease. Her lab is focused on the genomics of neurodevelopmental disorders, optimization of genomic workflows, and application of novel genomic technologies to understand disease. The research is presented in De Novo Variant Calling Identifies Cancer Mutation Signatures in the 1000 Genomes Project, published in Human Mutation.
Dr. Turner worked closely with the NVIDIA genomic team to integrate her trio analysis into NVIDIA Clara Parabricks. Dr. Turner was astonished to see a 100x speedup in turnaround time for a trio analysis using NVIDIA Clara Parabricks. The initial analysis to generate DNVs on GPUs took 8.5 hours using a server with just 4 GPUs versus 800 hours on CPUs. When the team further parallelized the workflow on GPUs, the run time was further shortened to less than one hour.
Dr. Turner has focused most of her career on DNVs, which are novel variants present in children’s DNA but not present in their parent’s DNA. These DNVs can be assessed by sequencing the DNA from a child and both parents followed by a comparative analysis, called a trio analysis. In the general population, each individual has around 40 to 100 DNVs and most DNVs do not affect the genes.
However, a genetic disease often results when a Single Nucleotide Variant (SNV) in a base pair (A,T, C, G), small insertion/deletion (indel), or Structural Variant (SV), alters a gene and affects the resulting protein production or function. This is the case with some neurodevelopmental disorders, where enrichment of protein-coding DNVs in patients has been identified in phenotypes including autism, epilepsy, intellectual disability, and congenital heart defects.
These fast results held promise not only for scientific discovery but also for Dr. Turner’s vision of same-day clinical results. To confirm the accuracy of the de novo variant calls from the new GPU-based workflow, the team leveraged NVIDIA Clara Parabricks to study a family with monozygotic twins, also known as identical twins, who have the same DNA.
The results showed the same number of DNVs in both the GPU-based and the previous CPU-based workflows, and in both cases about 20% CpG sites were found, indicating that the NVIDIA Clara Parabricks workflow produced equivalent results, but 100x faster. This meant that their autism genomic research could be completed faster, variants could be discovered faster, and hopefully insights for patients can be understood faster.
Dr. Turner remarked, “Utilization of GPUs is enabling rapid bioinformatic analyses to move forward to a one-hour genomic workup.”
With the new GPU-based DNV genomic analysis workflow, the team proceeded to study sequence data from the 1000 Genomes Project, an international research consortium that has sequenced representative cohorts from African, East Asian, South Asian, and European populations. The 1000 Genomes Project aims to describe and characterize the variations found in human genomes as a basis for investigating the relationship between genetic polymorphisms and phenotypes by sequencing 2,600 individuals from 26 populations from around the world.
Recently, The New York Genome Center sequenced these individuals at high depth and made the data publicly available. The population included 602 trios of families with no autism. This was the first opportunity to look at DNVs with no known phenotypes as a control to understand the level of DNVs in population and compare those to the autism cohort.
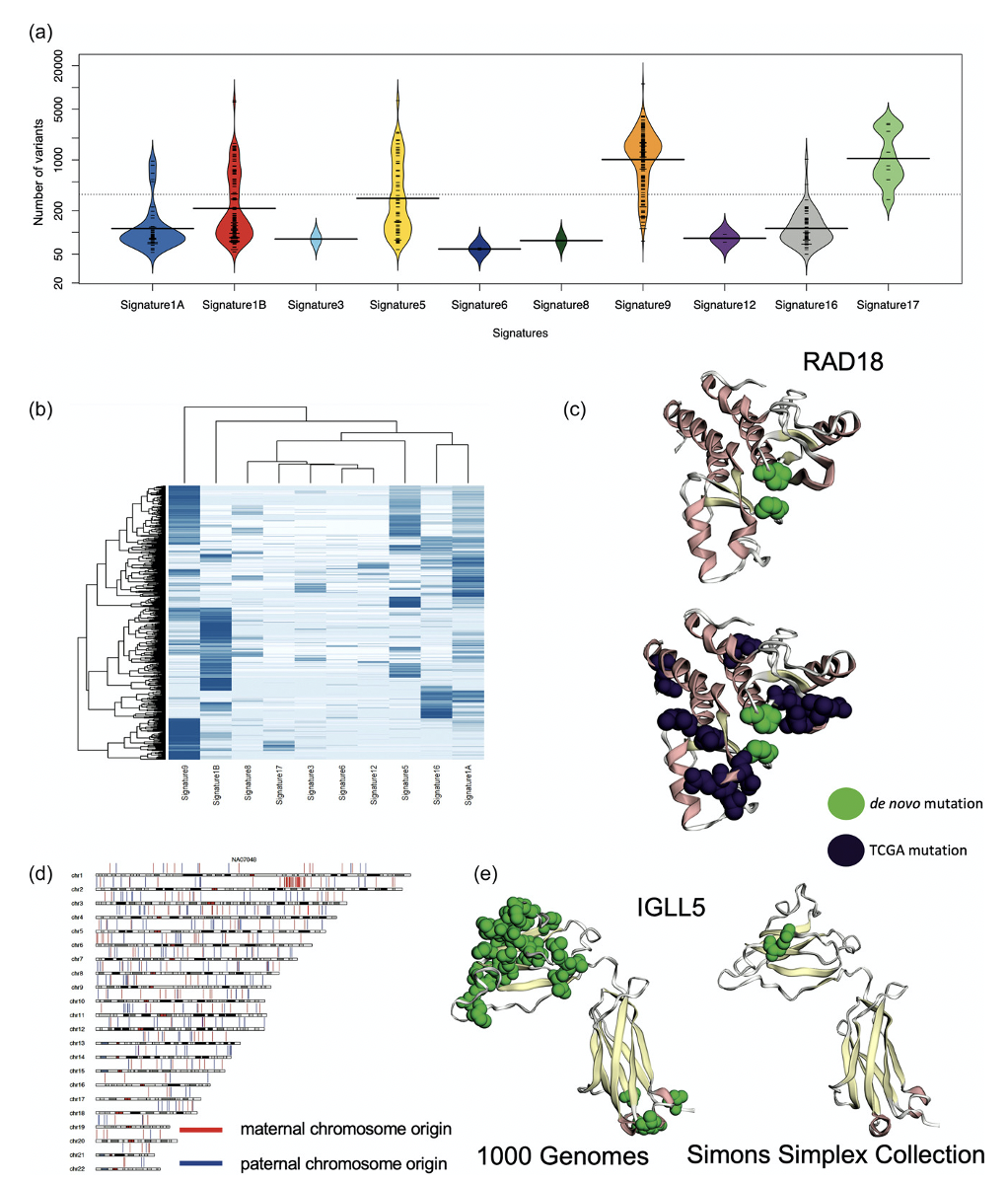
The DNV analysis of the 1000 Genomes Project individuals ended up surprising Dr. Turner’s team. They saw a bimodal distribution of the number of DNVs with peaks at 200, a little larger than expected, and at 2000, much larger than expected. Dr. Turner looked at the various cohorts in the 1000 Genomes Project data and noticed that the CEU population, which is a cohort of European individuals, has been studied for a longer time and therefore has been also cultured more, potentially leading to more cell line artifacts.
One individual, identified as NA12878 in the cohort, was sequenced multiple times: in 2012, 2013, twice in 2018, and in 2020. Dr. Turner showed that the DNVs had increased over time. 2020 had the most DNVs, supporting the conclusion that more cell line artifacts were in the 2020 samples versus the 2012 sample. The team concluded that although the 1000 Genomes Project is an excellent source of data for genomic study, it may not be ideal for filtering datasets for patient controls, due to the prevalence of cell line artifacts.
Though the 1000 Genomes Project provides critical biological and practical insights, only 20% of the children have the expected number of DNVs and considerable evidence indicates that excessive DNVs are cell line artifacts. The excess DNVs match mutation signatures of B-cell lymphoma cancers, demonstrating that cell line artifacts are not accumulating in a random manner.
Protein-coding DNVs are identified in DNA repair genes and may contribute to excess DNVs. The cohort of 602 individuals is significant for protein-coding DNVs in IGLL5 that is known to have excess mutations in B-cell lymphomas and individuals with these DNVs all have greater than 100 DNVs. Protein-coding DNVs are identified in clinically relevant variant sites warranting caution in using this data as a binary filtering set for patients. Future genomic studies performing genome sequencing should focus on either family-based approaches or utilized DNA derived directly from blood for building good controls and reference data bases.
Dr. Turner commented, “My lab was excited to develop a de novo variant calling workflow that utilizes GPUs which enabled us to quickly analyze nearly 4,800 whole-genome sequenced parent-child trios to gain important biological insights.”
An accelerated suite of tools to power genomic research
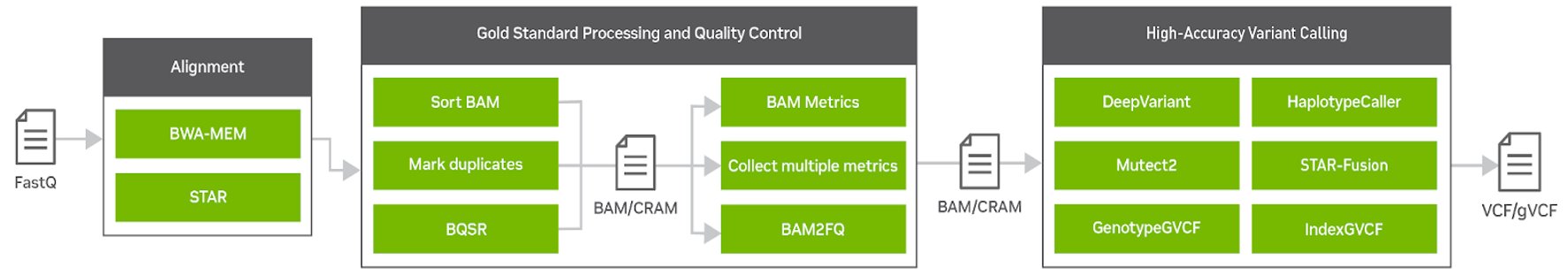
Clara Parabricks v4.0 is a more focused genomic analysis toolset than previous versions, with rapid alignment, gold standard processing, and high accuracy variant calling. It offers the flexibility to freely and seamlessly intertwine GPU and CPU tasks and prioritize the GPU-acceleration of the most popular and bottlenecked tools in the genomics workflow. Clara Parabricks can also integrate cutting-edge deep learning approaches in genomics.
You can register to download Clara Parabricks for free. You can also request a free Clara Parabricks NVIDIA LaunchPad Lab demo to experience accelerated industry-standard tools for germline and somatic analysis for an exome and whole genome dataset.
For more information about Clara Parabricks, including technical details on the tools available, check out the Clara Parabricks documentation.
