Creating new drug candidates is a heroic endeavor, often taking over 10 years to bring a drug to market. New supercomputing-scale large language models (LLMs) that understand biology and chemistry text are helping scientists understand proteins, small molecules, DNA, and biomedical text.
These state-of-the-art AI models help generate de novo proteins and molecules and predict the 3D structures of proteins. They can predict the binding structure of a small molecule to a protein and are offering scientists easier ways to engineer new candidate drugs and ultimately bring hope for patients.
After Exscientia brought an AI-designed drug candidate to a clinical trial in 2021, several other companies have announced that their candidates are in trials. Within drug companies focused on AI-based discovery, there is publicly available information on about 160 discovery programs, of which 15 products are reportedly in clinical development.
At the forefront of AI-based drug discovery are generative AI models for applications such as generating high-quality proteins. These large, powerful models learn from unlabeled data (such as sequencing data) on multi-GPU, multi-node, high-performance computing (HPC) infrastructures.
With NVIDIA BioNeMo Service, workflows for generative AI for biology are optimized and turnkey. You can focus on adapting AI models to the right drug candidates instead of dealing with configuration files and setting up supercomputing infrastructure.
BioNeMo Service
BioNeMo Service is a cloud service for generative AI in early drug discovery, featuring nine state-of-the-art large language and diffusion models in one place.
The models in BioNeMo are accessible through a web interface or fully managed APIs and can be further trained and optimized on NVIDIA DGX Cloud.
With BioNeMo Service, you can perform any of the following tasks:
- Generate large libraries of proteins.
- Build property predictors using embeddings to refine protein libraries.
- Generate small molecules with specific properties.
- Rapidly and accurately predict and visualize the 3D structure for billions of proteins.
- Run large campaigns of ligand-to-small-molecule pose estimations.
- Download proteins, molecules, and predicted 3D structures.
Generative AI models in BioNeMo Service
BioNeMo Service features nine AI generative models covering a wide spectrum of applications for developing AI drug discovery pipelines:
- AlphaFold 2, ESMFold, and OpenFold for 3D protein structure prediction from a primary amino acid sequence
- ESM-1nv and ESM-2 for protein property predictions
- ProtGPT2 for protein generation
- MegaMolBART and MoFlow for small molecule generation
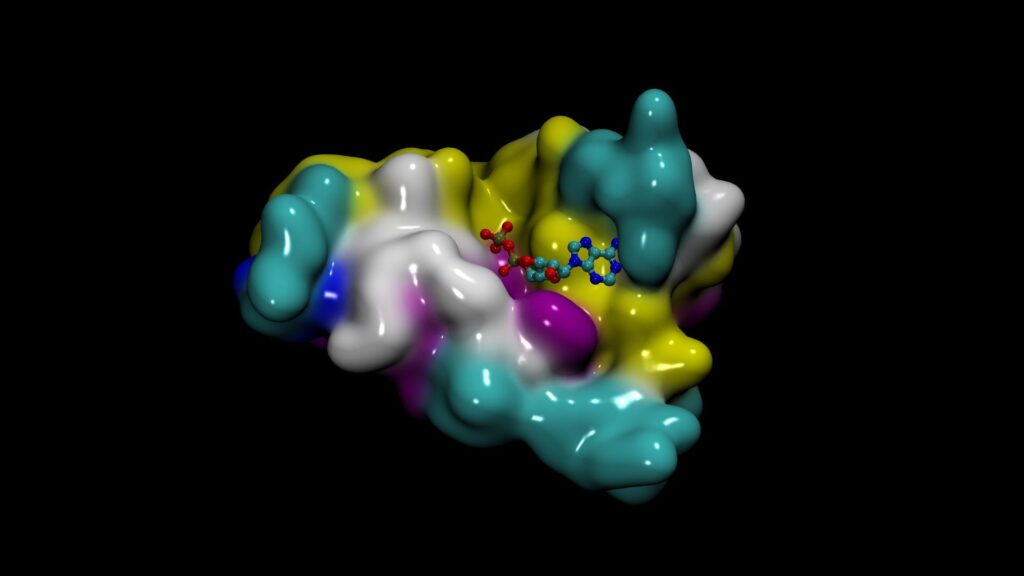
- DiffDock for predicting the binding structure of a small molecule to a protein
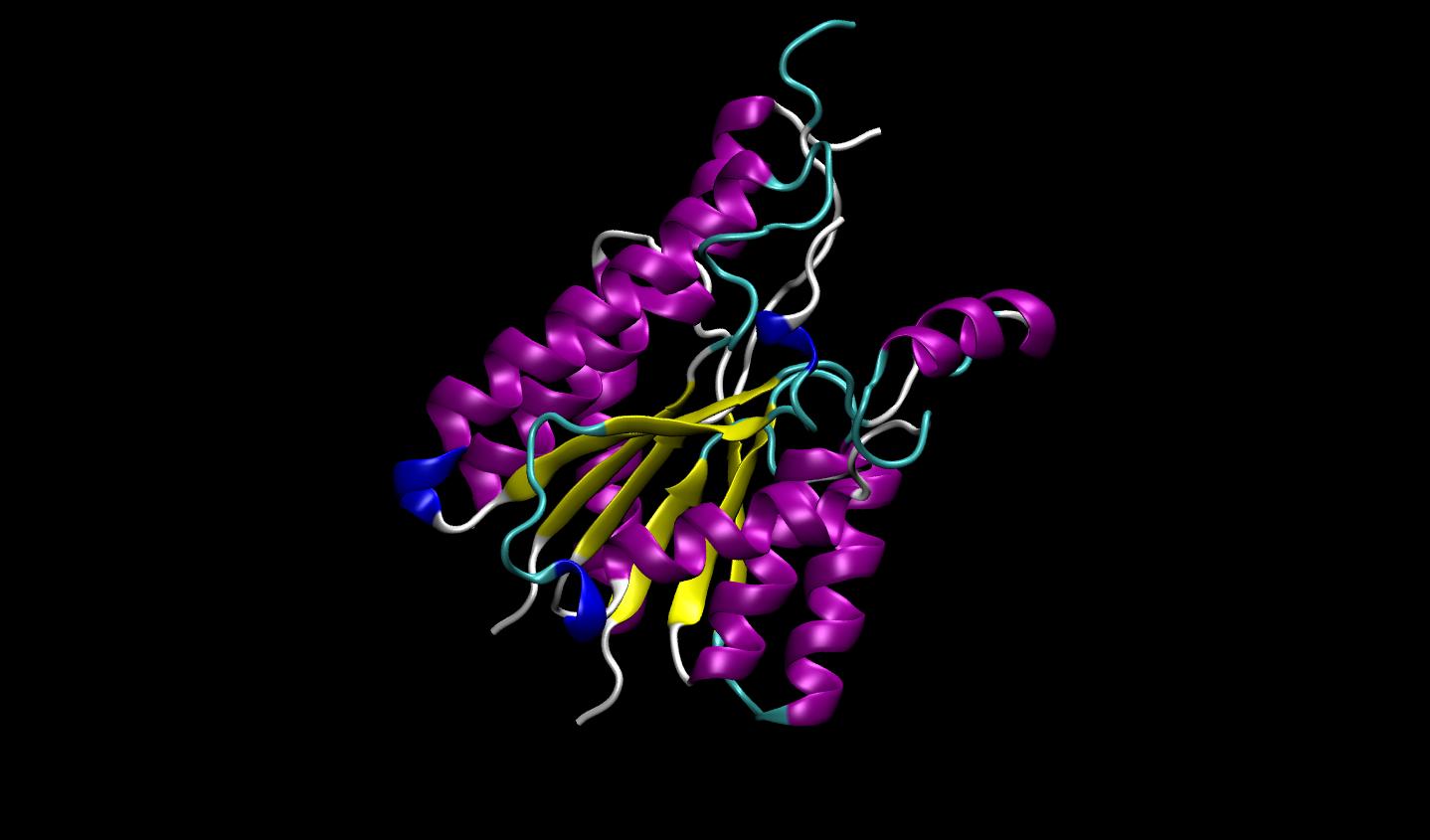
3D protein structure prediction
Protein structure prediction models enable scientists to predict the 3D protein structure from its primary linear amino acid sequence. AlphaFold 2, OpenFold, and ESMFold are models available in BioNeMo Service for protein structure prediction.
DeepMind’s AlphaFold 2 reached a significant milestone at CASP14, where it achieved near experimental accuracy for predicted protein 3D structures. Using deep learning and developed in JAX, AlphaFold 2 predicts the relationship between amino acid sequences of a protein and its 3D structures with high accuracy, even when only a few homologous sequences are available.
OpenFold is a faithful reproduction of DeepMind’s AlphaFold 2 model for 3D protein structure prediction from a primary amino acid sequence. While AlphaFold 2 was developed in a JAX workflow, OpenFold bases its code on PyTorch. OpenFold achieves similar accuracy to the original model and predicts the median backbone at an accuracy of 0.96 Å RMSD95.
BioNeMo accelerates OpenFold by 6x so that drug discovery researchers can analyze larger datasets and perform more iterations. OpenFold in BioNeMo is also trainable, meaning that variants may be created for specialized research.
Meta’s ESMFold is a transformer-based, ultrafast, 3D protein structure prediction based on ESM-2 embeddings without multiple sequence alignment (MSA). It includes a folding head to enable a fully end-to-end, single-sequence structure predictor. The model learns protein sequences through unsupervised pretraining and can predict the structure of a single protein sequence without requiring many homologous sequences as input.
On a single NVIDIA GPU, ESMFold makes a prediction of a protein with 384 residues in 14.2 seconds, 6x faster than a single AlphaFold 2 model. On shorter sequences, the improvement increases up to ∼60x.
Protein property predictions
Meta’s state-of-the-art ESM-1 and ESM-2 are LLMs for the evolutionary-scale modeling of proteins. They are inspired by the BERT architecture and trained on millions of protein sequences with a masked language modeling objective. ESM-1 and ESM-2 learned the patterns and dependencies between amino acids that ultimately give rise to 2D protein structure and function.
ESM-1nv in BioNeMo is a faithful reproduction of Meta’s ESM-1b, which is an LLM for the evolutionary-scale modeling of proteins. It is based on the BERT architecture and trained on millions of protein sequences with a masked language modeling objective. ESM-1nv learns the patterns and dependencies between amino acids that ultimately give rise to protein structure and function.
Through BioNeMo, ESM-1nv is optimized to be re-trainable, extensible, and fine-tunable on large compute infrastructures, such as DGX Cloud.
Where ESM-1nv is typically used to predict multiple protein properties from amino acid sequences, ESM-2 is typically used to predict the effects of mutations on protein stability.
Embeddings from ESM-1nv and ESM-2 in BioNeMo Service can be used to fit downstream task models for protein properties of interest, such as subcellular location, thermostability, water solubility, and conserved regions or variable regions. This is accomplished by training a typically much smaller model with a supervised learning objective to infer a property from the embeddings of protein sequences. This approach has been shown to provide state-of-the-art accuracy on a range of prediction tasks.
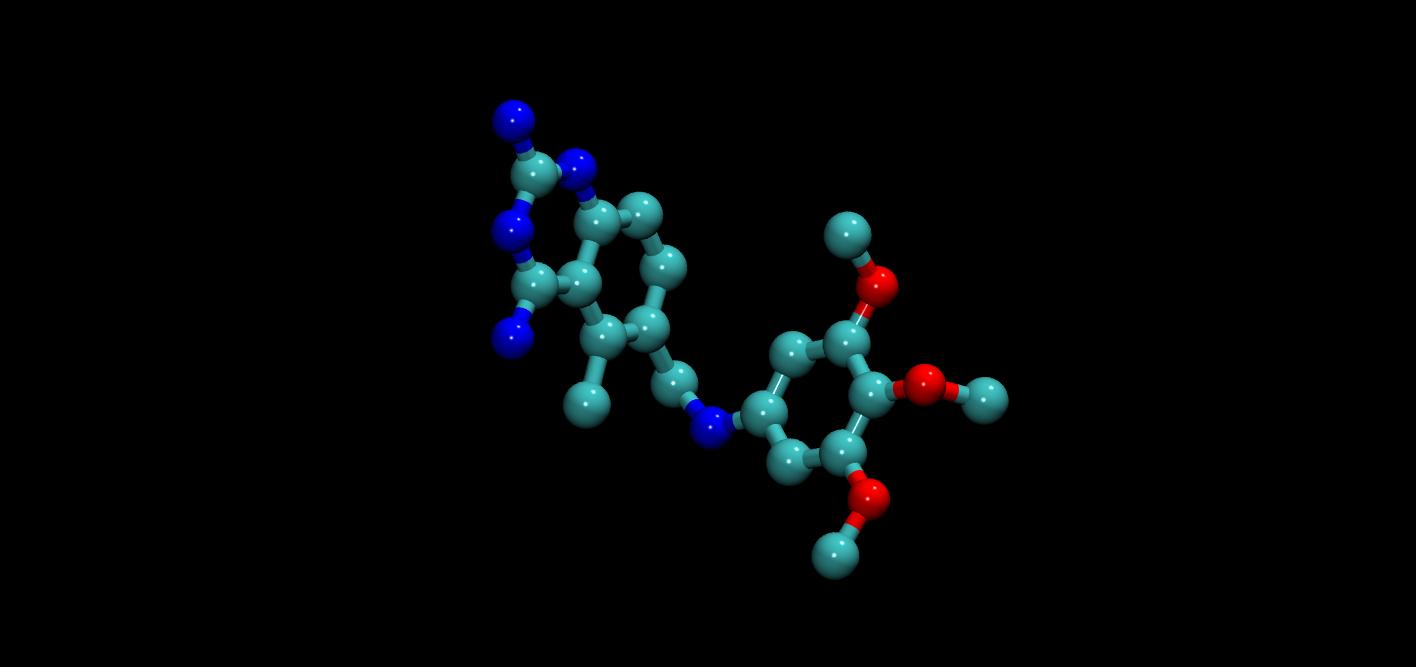
Small molecule generation
MegaMolBART and MoFlow are generative chemistry AI models in BioNeMo. MegaMolBart is a large, transformer-based generative chemistry model used for molecular optimization. It uses SMILES, a string notation for representing the chemical structure of small molecules. Developed by AstraZeneca and NVIDIA, MegaMolBART is optimal for generating new small molecules with binding affinities that have been tested experimentally, as well as for molecule embeddings.
MegaMolBART relies on NVIDIA NeMo, which provides a robust environment for developing, training, and deploying deep learning models, including Megatron models.
NeMo provides enhancements to PyTorch Lighting such as hyperparameter configurability with YAML files and checkpoint management. It also enables the development and training of large transformer models using NVIDIA NeMo-Megatron, which makes multi-GPU, multi-node training with data parallelism, model parallelism, and mixed precision easily configurable.
The ZINC-15 database was used for pretraining MegaMolBART. Approximately 1.45B molecules (SMILES strings) were selected from tranches meeting the following constraints:
- Molecular weight <= 500 Daltons
- LogP <= 5
- Reactivity level was “reactive”
- Purchasability was “annotated”
MoFlow, a flow-based generative model from a team at Weill Cornell Medicine, learns invertible mappings between molecular graphs and their latent representations. Generating molecular graphs with desired chemical properties driven by deep graph generative models can accelerate the drug discovery process.
MoFlow achieves state-of-the-art performance with a novel, conditional flow–based approach. It uses graph convolutions and produces a valid molecular graph that follows bond-valence constraints. It is used for molecule generation, reconstruction, and optimization.
Protein generation
ProtGPT2, created at ISMB and the Universität of Bayreuth, Germany, is an LLM based on the GPT2 transformer architecture that generates de novo protein sequences to identify unique structures, properties, and functions. The model is optimal for generating custom protein sequences when there is limited training data.
It is trained on the protein space database UniRef50 and has 36 layers with 738M parameters. ProtGPT2 was trained using a causal modeling objective, in which the model is trained to predict the next token (or, in this case, oligomer) in the sequence. By doing so, the model learns an internal representation of proteins and can speak the protein language.
Molecular docking
DiffDock from MIT’s Jameel Clinic is a diffusion generative AI model. It predicts the binding structure of a small molecule ligand to a protein, known as molecular docking or pose prediction.
DiffDock has fast inference times and provides confidence estimates with high selective accuracy. The model is highly accurate and computationally efficient. It achieved a new state-of-the-art 38% top-1 prediction with RMSD<2A on the PDBBind blind docking benchmark, considerably surpassing the previous best search-based (23%) and deep learning methods (20%).
DiffDock was evaluated on molecular complexes from the PDBBind benchmark and compared with state-of-the-art search-based methods such as SMINA and GLIDE, as well as recent deep learning methods EquiBind and TANKBind. DiffDock can help AI drug discovery pipelines and open new research avenues for downstream task integrations.
Get started with BioNeMo
To learn more about BioNeMo and apply for early access to BioNeMo Service, see the BioNeMo page.
Taking place this week, NVIDIA GTC 2023 has numerous sessions on the latest AI advances in drug discovery. Register free to get access to all the content on-demand and check out the following sessions on AI drug discovery and BioNeMo:
- A Transformative AI Platform to Accelerate Biologics Discovery
- Generative Deep Learning with BioNeMo for Protein Therapeutics
- AI-Powered Drug Discovery
- Understanding the Chemical and Biological Language of Life with LLMs using BioNeMo
- Using AI to Accelerate Scientific Discovery
- Artificial Intelligence Captures the Language of Life Written in Proteins
