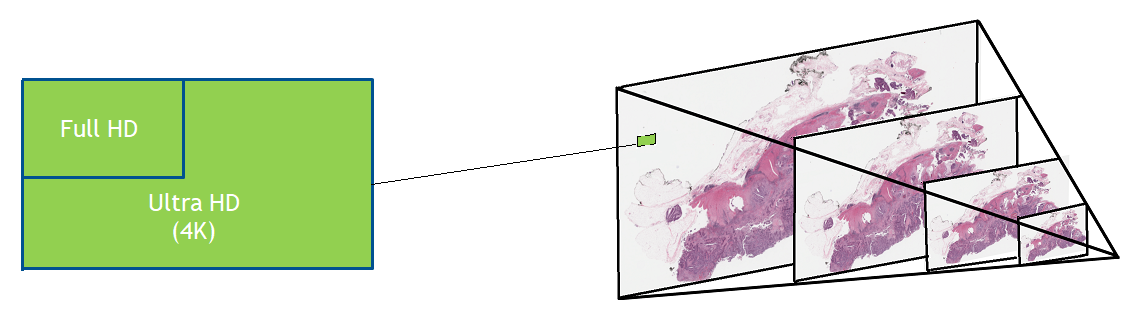
数字病理切片扫描仪生成大量图像。载玻片通常以 40 倍的放大率进行扫描,得到千兆像素的图像。压缩可以将每个幻灯片的文件大小减少到 1 或 2 GB ,但这种数据量在移动、保存、加载和查看方面仍然具有挑战性。要以全分辨率查看典型的完整幻灯片图像,需要一个网球场大小的监视器。
与组织病理学一样,基因组学和显微镜可以产生数兆字节的数据。有些用例涉及多种模式,将这些数据转换为更易于管理的大小通常需要进行渐进式转换,直到只保留最显著的特征。本文探讨了实现这种数据细化的一些方法,使用的分析类型,以及诸如MONAI和RAPIDS可以释放有意义的见解。以一个典型的数字组织病理学图像为例,因为这些图像现在在全球的常规临床环境中使用。
MONAI 是一套开源、免费的协作框架,旨在加速医学成像领域的研究和临床协作。 RAPIDS 是一套开源软件库,用于在 GPU 上构建端到端的数据科学和分析管道。RAPIDS cuCIM 是一个用于多维图像的计算机视觉处理软件库,可以加速 MONAI 的成像,以及cuDF library 可以帮助完成工作流所需的数据转换。
管理整个幻灯片图像数据
以前的研究表明cuCIM 可以加快整个幻灯片图像的加载速度。例如,使用 cuCIM 加速 Scikit-Image API:在 GPU 上进行 n 维图像处理和 I/O。
但是,管道的其余部分呢,可能包括图像预处理、推理、后处理、可视化和分析?越来越多的仪器捕捉各种数据,包括多光谱图像、遗传和蛋白质组学数据,所有这些都面临着类似的挑战。
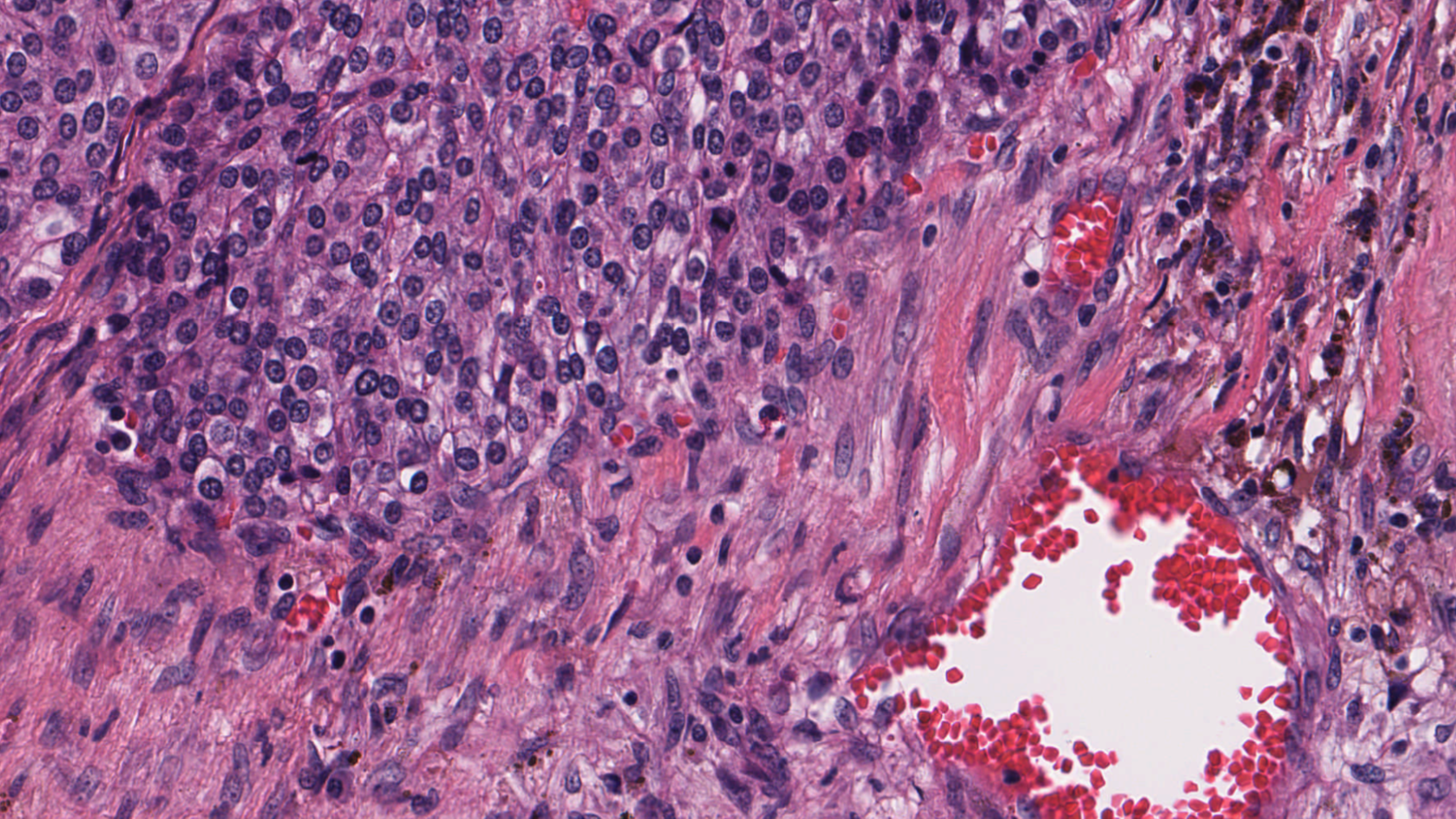
像癌症这样的疾病来自细胞核,细胞核只有约 5-20 微米大小。为了辨别各种细胞亚型,病理学家需要看到其形状、颜色、内部纹理和模式。这需要非常大的图像。
考虑到 2D 深度学习算法(如 DenseNet )的常见输入大小通常约为 200 x 200 像素,高分辨率图像只需一张幻灯片就需要分割成补丁——可能为 100000 个。
玻片制备和组织染色过程可能需要数小时。虽然低延迟推理结果的价值很小,但分析仍必须跟上数字扫描仪的采集率,以防止积压。因此,吞吐量至关重要。提高吞吐量的方法是更快地处理图像或同时计算多个图像。
潜在解决方案
数据科学家和开发人员已经考虑了许多方法来使问题更容易处理。考虑到图像的大小和病理学家进行诊断的时间有限,没有实际的方法来以全分辨率查看每个像素。
相反,他们以较低的分辨率查看图像,然后放大他们确定的可能包含感兴趣特征的区域。他们通常可以在观看了 1-2% 的全分辨率图像后做出诊断。在某些方面,这就像犯罪现场的侦探:现场的大部分内容都无关紧要,结论通常取决于提供关键信息的一两个纤维或指纹。
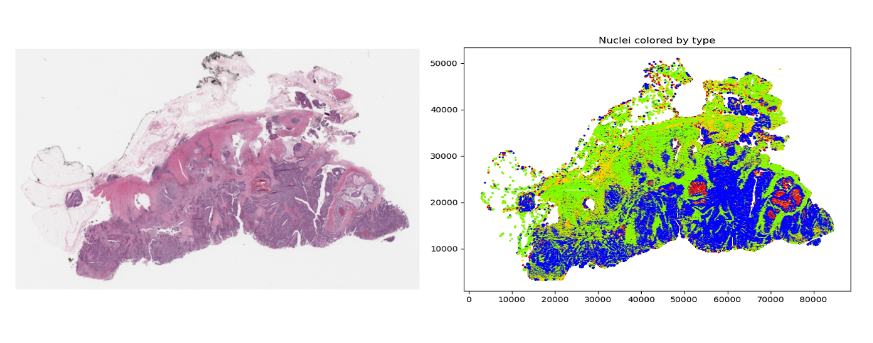
与人类同行不同,人工智能和机器学习( ML )无法丢弃图像中 98-99% 的像素,因为担心它们可能会错过一些关键细节。这在未来可能是可能的,但需要相当大的信任和证据来证明它是安全的。
在这方面,当前的算法对所有输入像素一视同仁。各种算法机制随后可以为它们分配或多或少的权重(注意力、最大池、偏差和权重),但最初它们都具有影响预测的相同潜力。
这不仅给组织病理学处理管道带来了巨大的计算负担,而且还需要在磁盘 CPU 和 GPU 之间移动大量数据。大多数组织病理学幻灯片包含空白、冗余信息和噪音。可以利用这些特性来减少提取重要信息所需的实际计算。
例如,病理学家对相关区域内的某些细胞类型进行计数以对疾病进行分类可能就足够了。要做到这一点,该算法必须将像素强度值转换为具有相关细胞类型标签的细胞核质心阵列。因此,计算一个区域内的细胞计数非常简单。有许多方法可以将整个幻灯片图像过滤为特定任务的基本元素。一些例子可能包括:
MONAI 和 RAPIDS
对于这些方法中的任一种,MONAI 提供了多种模型和训练管道,您可以根据自己的需求进行自定义。大多数模型都是通用的,可以满足数据的特定要求(如通道数量和维度),但也有一些是特定的,比如数字病理学。
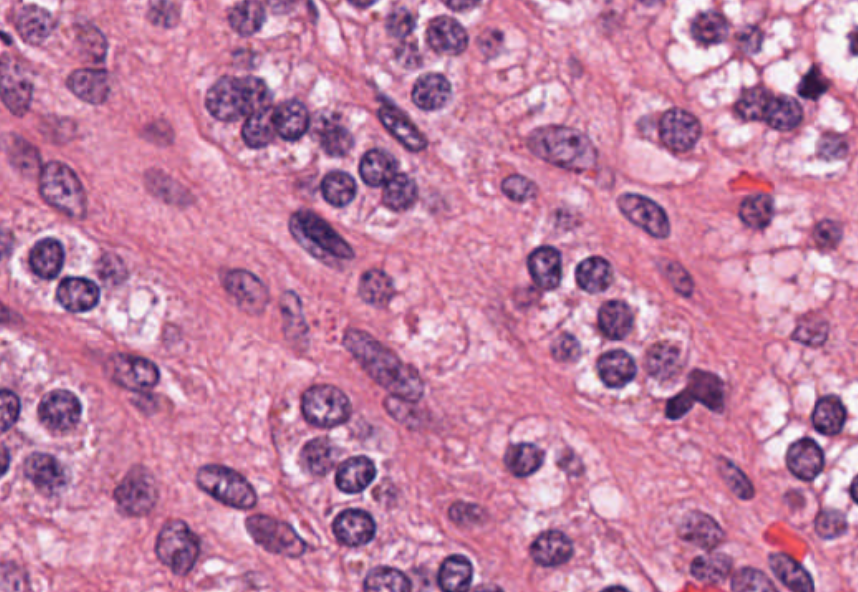
一旦导出了这些特征,就可以将其用于分析。然而,即使在这种类型的降维之后,仍可能有许多特征需要分析。例如,图 3 显示了一张包含数十万个细胞核的图像(最初为 100K x 60K RGB 像素)。即使为每个 64 x 64 瓦片生成嵌入,仍然可能为一张幻灯片生成数百万个数据点。
这就是RAPIDS可以提供的帮助。它是一个开源 GPU 库套件,使用 Python 加速数据科学,包括涵盖一系列常见活动的工具,如机器学习(ML)、图形分析、ETL 和可视化。有一些底层技术,例如CuPy,它使不同的操作能够访问 GPU 存储器中的相同数据,而无需复制或重构底层数据。这是 RAPIDS 如此快速的主要原因之一。
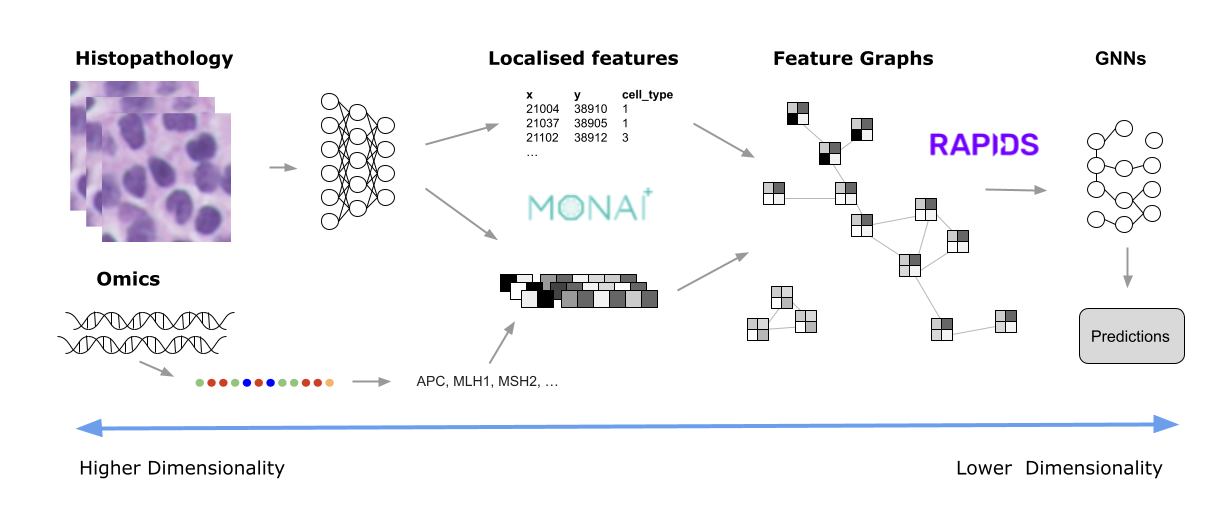
开发人员的主要交互工具之一是 CUDA 加速的数据帧(cuDF)。数据以表格形式显示,可以使用cuDF API,具有类似 pandas 的命令,易于采用。然后,这些数据帧被用作许多其他 RAPIDS 工具的输入。
例如,假设您想从所有核创建一个图,将每个核连接到某个半径内的最近邻居。为此,您需要向 cuGraph API 提供表示每个图边缘的源节点和目标节点的列(具有可选权重)。要生成此列表,可以使用 cuML 的最近邻居搜索功能。只需提供一个列出所有核坐标的数据帧,cuML 就可以完成所有繁重的工作。
from cuml.neighbors import NearestNeighbors
knn = NearestNeighbors()
knn.fit(cdf)
distances, indices = knn.kneighbors(cdf, 5)请注意,默认情况下,计算的距离是欧几里得距离,为了节省不必要的计算,它们是平方值。其次,该算法可以默认使用启发式。如果需要实际值,可以指定可选的algorithm=‘brute’参数无论哪种方式,在 GPU 上的计算都非常快。
接下来,将距离和索引数据帧合并为一个单独的数据帧。为此,需要首先为距离列指定唯一的名称:
distances.columns=['ix2','d1','d2','d3','d4']
all_cols = cudf.concat(
[indices[[1,2,3,4]], distances[['d1','d2','d3','d4']]],
axis=1)
每一行都必须对应于图中的一条边,因此需要将数据帧拆分为每个最近邻居的一行。然后,这些列可以重命名为“ source ”、“ target ”和“ distance ”
all_cols['index1'] = all_cols.index
c1 = all_cols[['index1',1,'d1']]
c1.columns=['source','target','distance']
c2 = all_cols[['index1',2,'d2']]
c2.columns=['source','target','distance']
c3 = all_cols[['index1',3,'d3']]
c3.columns=['source','target','distance']
c4 = all_cols[['index1',4,'d4']]
c4.columns=['source','target','distance']
edges = cudf.concat([c1,c2,c3,c4])
edges = edges.reset_index()
edges = edges[['source','target','distance']]消除所有超过一定距离的邻居,请使用以下过滤器:
distance_threshold = 15
edges = edges.loc[edges["distance"] < distance_threshold**2]在这一点上,您可以省去“距离”列,除非图中的边需要加权。然后创建图形本身:
cell_graph = cugraph.Graph()
cell_graph.from_cudf_edgelist(edges,source='source', destination='target', edge_attr='distance', renumber=True)
有了图形之后,就可以进行标准的图形分析操作了。三角形计数是长度为三的循环数。图的 k 核是包含 k 度或更高阶节点的极大子图:
count = cugraph.triangle_count(cell_graph)
coreno = cugraph.core_number(cell_graph)
也可以可视化图形,即使它可能包含数十万条边。使用现代 GPU,可以实时查看和导航图形。要生成这样的可视化效果,请使用 cuXFilter:
nodes = tiles_xy_cdf
nodes['vertex']=nodes.index
nodes.columns=['x','y','vertex']
cux_df = fdf.load_graph((nodes, edge_df))
chart0 = cfc.graph(
edge_color_palette=['gray', 'black'],
timeout=200,
node_aggregate_fn='mean',
node_pixel_shade_type='linear',
edge_render_type='direct',#other option available -> 'curved', edge_transparency=0.5)
d = cux_df.dashboard([chart0], layout=clo.double_feature)
chart0.view()
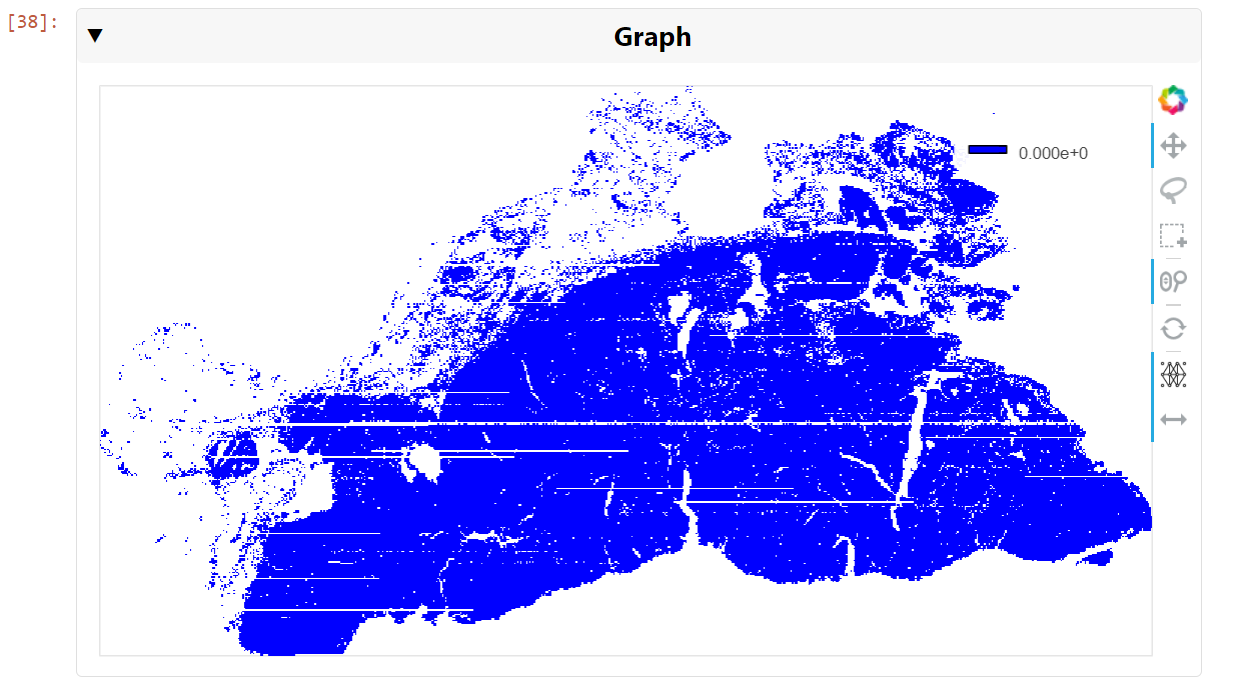
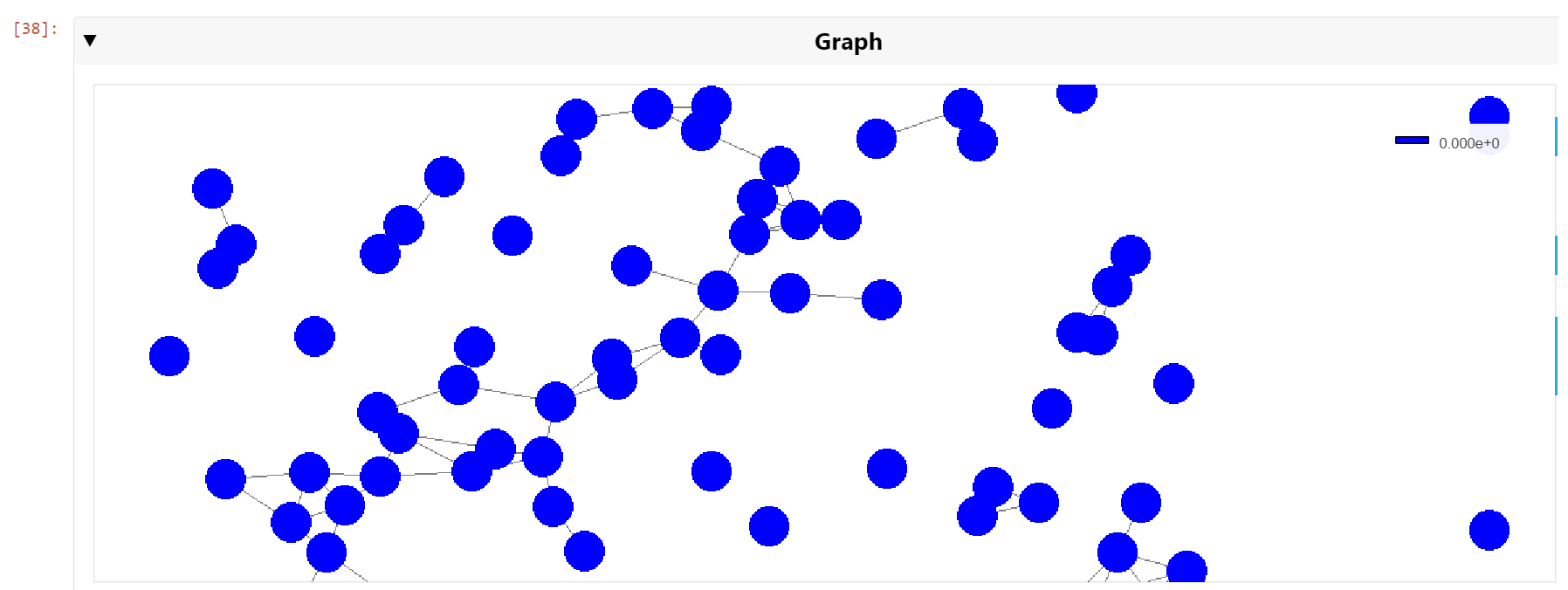
然后,您可以平移并缩小到细胞核级别,以查看最近邻居的集群(图 6 )。
结论
从原始像素中提取见解可能既困难又耗时。一些强大的工具和技术可以应用于大型图像问题,即使是最具挑战性的数据也可以提供近乎实时的分析。除了机器学习功能,GPU-RAPIDS等加速工具也提供了强大的可视化功能,有助于解读基于深度学习的方法产生的计算特征。本文描述了一套端到端的工具,可以使用深度学习、机器学习图和图神经网络方法。
使用RAPIDS和MONAI,在您的数据上释放 GPU 的力量。加入MONAI 社区,在 NVIDIA 开发者论坛上参与讨论。