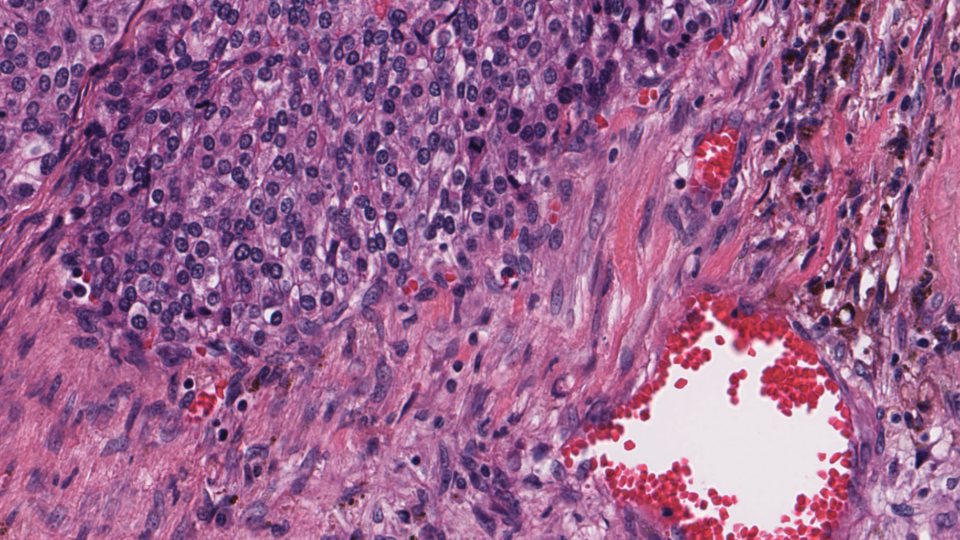
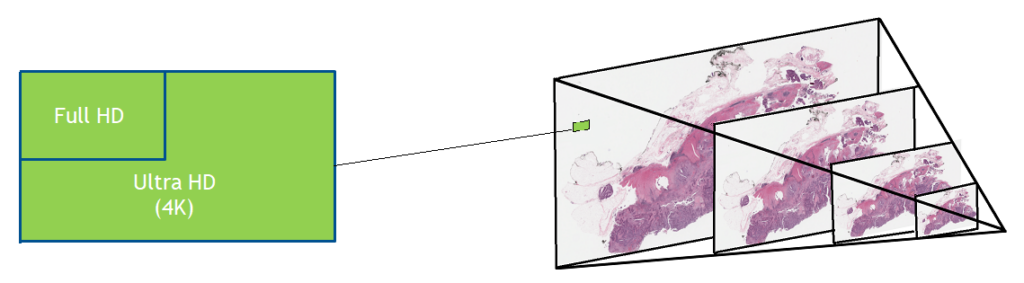
디지털 병리 슬라이드 스캐너는 방대한 이미지를 생성합니다. 유리 슬라이드는 일반적으로 40배율로 스캔되어 기가픽셀 이미지가 생성됩니다. 압축을 통해 파일 크기를 슬라이드당 1~2GB로 줄일 수 있지만, 이 정도 용량의 데이터는 이동, 저장, 불러오기, 보기가 여전히 어렵습니다. 일반적인 전체 슬라이드 이미지를 전체 해상도로 보려면 테니스 코트 크기 정도의 모니터가 필요합니다.
조직 병리학과 마찬가지로 유전체학 및 현미경 검사 모두 테라바이트 단위의 데이터를 쉽게 생성할 수 있습니다. 일부 사용 사례에는 여러 모달리티가 포함됩니다. 이러한 데이터를 관리하기 쉬운 크기로 변환하려면 일반적으로 가장 두드러진 특징만 남을 때까지 점진적인 변환이 필요합니다. 이 게시물에서는 이러한 데이터 정제 작업을 수행할 수 있는 몇 가지 방법, 사용되는 분석 유형, MONAI 및 RAPIDS와 같은 도구로 의미 있는 인사이트를 얻는 방법에 대해 살펴봅니다. 현재 전 세계의 일상적인 임상 환경에서 사용되는 일반적인 디지털 조직 병리 이미지를 예로 들어 설명합니다.
MONAI는 의료 영상 분야의 연구 및 임상 협업을 가속화하는 데 최적화된 오픈 소스 무료 협업 프레임워크입니다. RAPIDS는 GPU에서 엔드투엔드 데이터 사이언스 및 분석 파이프라인을 구축하기 위한 오픈 소스 소프트웨어 라이브러리 모음입니다. 다차원 이미지를 위한 컴퓨터 비전 처리 소프트웨어 라이브러리인 RAPIDS cuCIM은 MONAI의 이미징을 가속화하고, cuDF 라이브러리는 워크플로우에 필요한 데이터 변환을 지원합니다.
전체 슬라이드 이미지 데이터 관리
이전 작업에서 cuCIM이 전체 슬라이드 이미지의 로딩 속도를 높이는 방법을 보여주었습니다. 예를 들어, cuCIM으로 Scikit-Image API 가속화하기: GPU에서 n차원 이미지 처리 및 I/O를 참조하세요.
하지만 이미지 전처리, 추론, 후처리, 시각화 및 분석을 포함하는 나머지 파이프라인은 어떻게 될까요? 다중 스펙트럼 이미지, 유전자 및 단백질체 데이터 등 다양한 데이터를 캡처하는 기기가 점점 더 많아지고 있으며, 모두 비슷한 과제를 안고 있습니다.
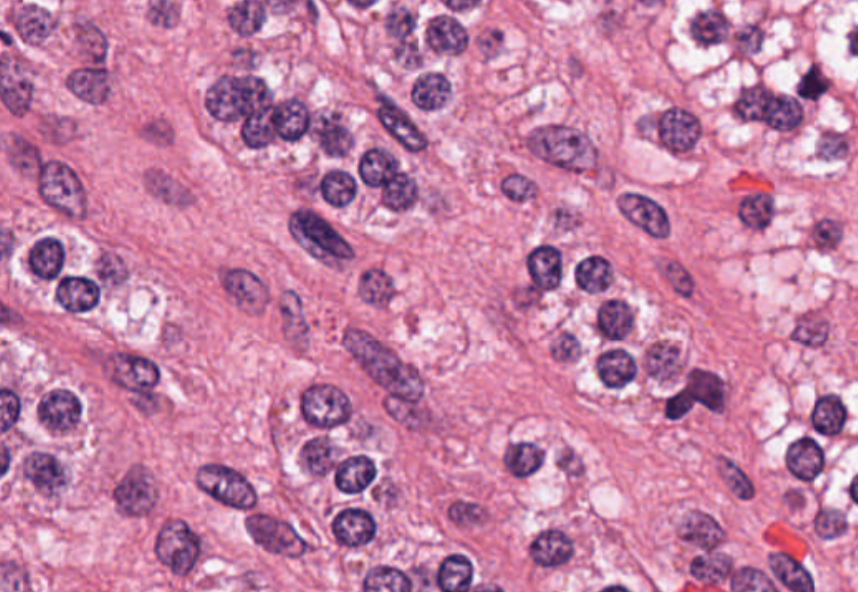
암과 같은 질병은 크기가 5~20미크론에 불과한 세포 핵에서 발생합니다. 다양한 세포 아형을 식별하려면 병리학자가 세포의 모양, 색상, 내부 질감, 패턴을 볼 수 있어야 합니다. 이를 위해서는 매우 큰 이미지가 필요합니다.
2D 딥러닝 알고리즘(예: DenseNet)의 일반적인 입력 크기가 약 200 x 200픽셀이라는 점을 감안할 때, 고해상도 이미지는 슬라이드 한 장을 위해 패치(잠재적으로 100,000개)로 분할해야 합니다.
슬라이드 준비와 조직 염색 과정에는 몇 시간이 걸릴 수 있습니다. 지연 시간이 짧은 추론 결과의 가치는 미미하지만, 백로그를 방지하기 위해 분석은 디지털 스캐너 획득 속도를 따라잡아야 합니다. 따라서 처리량이 중요합니다. 처리량을 개선하는 방법은 이미지를 더 빠르게 처리하거나 많은 이미지를 동시에 계산하는 것입니다.
잠재적 솔루션
데이터 사이언티스트와 개발자들은 이 문제를 해결하기 위해 여러 가지 접근 방식을 고려했습니다. 이미지의 크기와 병리학자가 진단을 내려야 하는 제한된 시간을 고려할 때, 모든 픽셀을 전체 해상도로 볼 수 있는 실용적인 방법은 없습니다.
대신 저해상도로 이미지를 검토한 다음 관심 있는 특징이 포함될 가능성이 있다고 판단되는 영역을 확대합니다. 일반적으로 전체 해상도 이미지의 1~2%만 보고도 진단을 내릴 수 있습니다. 어떤 면에서는 범죄 현장에 출동한 형사와 비슷합니다. 현장의 대부분은 관련이 없고, 핵심 정보를 제공하는 한두 개의 섬유나 지문에 의존해 결론을 내리는 경우가 많기 때문입니다.
인간과 달리 AI와 머신러닝(ML)은 중요한 디테일을 놓칠 수 있다는 우려 때문에 이미지 픽셀의 98~99%를 버릴 수 없습니다. 향후에는 가능할 수도 있지만, 안전하다는 것을 보여줄 수 있는 상당한 신뢰와 증거가 필요합니다.
이러한 측면에서 현재 알고리즘은 모든 입력 픽셀을 동일하게 처리합니다. 이후 다양한 알고리즘 메커니즘(주의, 최대 풀링, 바이어스 및 가중치)에 따라 더 많거나 적은 가중치를 부여할 수 있지만, 처음에는 모두 예측에 영향을 미칠 수 있는 동일한 잠재력을 가지고 있습니다.
이는 조직병리 처리 파이프라인에 큰 연산 부담을 줄 뿐만 아니라 디스크, CPU, GPU 간에 상당한 양의 데이터를 이동해야 합니다. 대부분의 조직병리 슬라이드에는 빈 공간, 중복 정보, 노이즈가 포함되어 있습니다. 이러한 특성을 악용하면 중요한 정보를 추출하는 데 필요한 실제 계산을 줄일 수 있습니다.
예를 들어, 병리학자가 질병을 분류하기 위해 해당 영역 내의 특정 세포 유형을 세는 것으로 충분할 수 있습니다. 이를 위해 알고리즘은 픽셀 강도 값을 관련 세포 유형 레이블이 있는 핵 중심 배열로 변환해야 합니다. 그러면 한 영역 내의 세포 수를 매우 간단하게 계산할 수 있습니다. 전체 슬라이드 이미지를 특정 작업에 필수적인 요소로 필터링하는 방법에는 여러 가지가 있습니다. 몇 가지 예를 들면 다음과 같습니다:
- 변형 자동 인코더 학습과 같은 비지도 방법을 사용하여 이미지 특징 집합을 학습하여 이미지 타일을 작은 임베딩으로 인코딩합니다.
- 관심 있는 모든 특징(예: 핵)을 로컬라이즈하고 이 정보만 사용하여 HoVerNet과 같은 전문 모델을 사용하여 메트릭을 도출합니다.
MONAI 및 RAPIDS
이러한 접근 방식 중 하나에 대해 MONAI는 필요에 따라 사용자 지정할 수 있는 다양한 모델과 훈련 파이프라인을 제공합니다. 대부분은 데이터의 특정 요구 사항(예: 채널 수 및 차원)에 맞게 조정할 수 있을 만큼 일반적이지만, 일부는 디지털 병리학과 같은 특정 분야에 특화되어 있습니다.
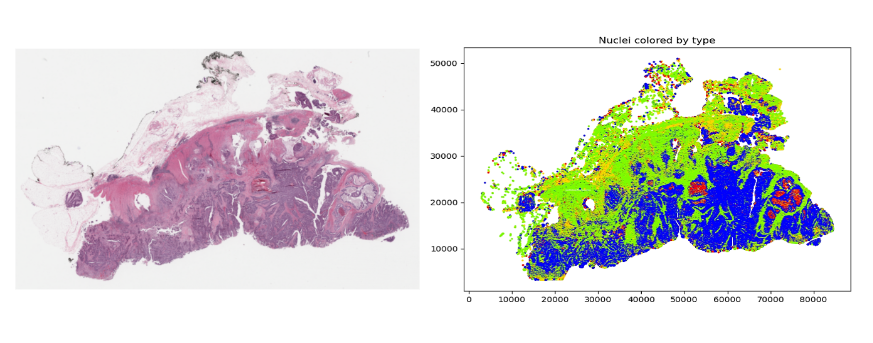
이러한 특징이 도출되면 분석에 사용할 수 있습니다. 그러나 이러한 유형의 차원 축소 후에도 분석해야 할 특징이 여전히 많이 있을 수 있습니다. 예를 들어, 그림 3은 수십만 개의 핵이 있는 이미지(원래는 100K x 60K RGB 픽셀)를 보여줍니다. 각 64 x 64 타일에 대한 임베딩을 생성하더라도 하나의 슬라이드에 수백만 개의 데이터 포인트가 발생할 수 있습니다.
이때 RAPIDS가 도움이 될 수 있습니다. Python으로 GPU 가속 데이터 과학을 위한 오픈 소스 라이브러리 제품군에는 ML, 그래프 분석, ETL 및 시각화와 같은 다양한 일반적인 활동을 다루는 도구가 포함되어 있습니다. 기본 데이터를 복사하거나 재구성하지 않고도 다양한 연산이 GPU 메모리의 동일한 데이터에 액세스할 수 있도록 하는 CuPy와 같은 몇 가지 기본 기술이 있습니다. 이것이 바로 RAPIDS가 빠른 속도를 자랑하는 주요 이유 중 하나입니다.
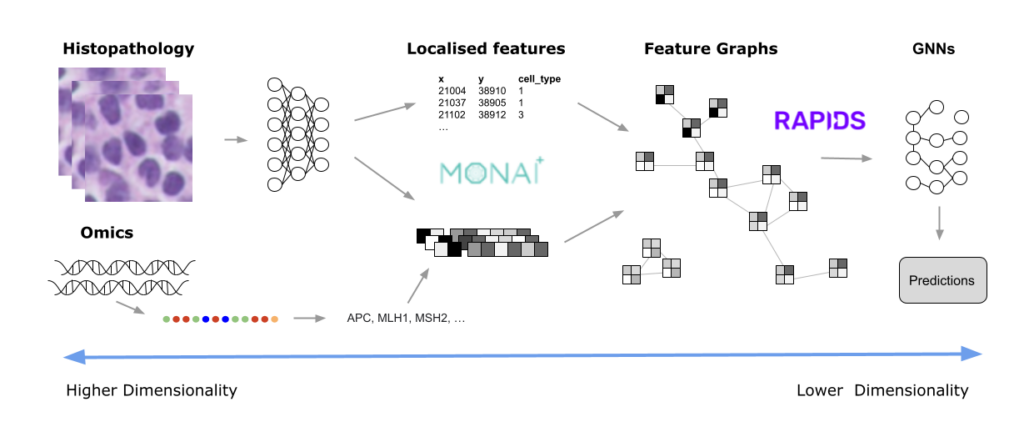
개발자를 위한 주요 인터랙션 도구 중 하나는 CUDA 가속 데이터프레임(cuDF)입니다. 데이터는 표 형식으로 표시되며, pandas와 유사한 명령어로 cuDF API를 사용하여 필터링하고 조작할 수 있으므로 쉽게 채택할 수 있습니다. 이러한 데이터 프레임은 다른 많은 RAPIDS 도구의 입력으로 사용됩니다.
예를 들어, 모든 핵으로부터 각 핵을 특정 반경 내의 가장 가까운 이웃과 연결하는 그래프를 만들고 싶다고 가정해 보겠습니다. 이렇게 하려면 각 그래프 엣지의 소스 및 대상 노드를 나타내는 열(선택적 가중치 포함)이 있는 데이터 프레임을 cuGraph API에 제공해야 합니다. 이 목록을 생성하려면 cuML 가장 가까운 이웃 검색 기능을 사용할 수 있습니다. 다시 말하지만, 모든 핵 좌표가 나열된 데이터 프레임을 제공하기만 하면 cuML이 모든 작업을 수행합니다.
from cuml.neighbors import NearestNeighbors
knn = NearestNeighbors()
knn.fit(cdf)
distances, indices = knn.kneighbors(cdf, 5)계산된 거리는 기본적으로 유클리드 거리이며, 불필요한 계산을 줄이기 위해 제곱된 값이라는 점에 유의해야합니다. 둘째, 알고리즘은 기본적으로 휴리스틱을 사용할 수 있습니다. 실제 값을 원하면 algorithm=‘brute’ 매개변수(선택 사항)를 지정할 수 있습니다. 어느 쪽이든 GPU에서 계산이 매우 빠릅니다.
다음으로 거리와 인덱스 데이터 프레임을 하나의 단일 데이터 프레임으로 병합합니다. 이렇게 하려면 먼저 거리 열에 고유한 이름을 할당해야 합니다:
distances.columns=['ix2','d1','d2','d3','d4']
all_cols = cudf.concat(
[indices[[1,2,3,4]], distances[['d1','d2','d3','d4']]],
axis=1)각 행은 그래프의 가장자리에 해당해야 하므로 데이터 프레임을 가장 가까운 각 이웃에 대해 행으로 분할해야 합니다. 그런 다음 열의 이름을 ‘source’, ‘target’, ‘distance’로 바꿀 수 있습니다.
all_cols['index1'] = all_cols.index
c1 = all_cols[['index1',1,'d1']]
c1.columns=['source','target','distance']
c2 = all_cols[['index1',2,'d2']]
c2.columns=['source','target','distance']
c3 = all_cols[['index1',3,'d3']]
c3.columns=['source','target','distance']
c4 = all_cols[['index1',4,'d4']]
c4.columns=['source','target','distance']
edges = cudf.concat([c1,c2,c3,c4])
edges = edges.reset_index()
edges = edges[['source','target','distance']]특정 거리를 초과하는 모든 이웃을 제거하려면 다음 필터를 사용합니다:
distance_threshold = 15
edges = edges.loc[edges["distance"] < distance_threshold**2]이 시점에서 그래프 내의 가장자리에 가중치를 부여할 필요가 없다면 ‘거리’ 열을 생략할 수 있습니다. 그런 다음 그래프 자체를 만듭니다:
cell_graph = cugraph.Graph()
cell_graph.from_cudf_edgelist(edges,source='source', destination='target', edge_attr='distance', renumber=True)그래프를 얻은 후에는 표준 그래프 분석 작업을 수행할 수 있습니다. 삼각형 수는 길이 3의 사이클 수입니다. 그래프의 k-코어는 차수 k 이상의 노드를 포함하는 최대 하위 그래프입니다:
count = cugraph.triangle_count(cell_graph)
coreno = cugraph.core_number(cell_graph)그래프에 수십만 개의 에지가 포함되어 있어도 그래프를 시각화할 수 있습니다. 최신 GPU를 사용하면 그래프를 실시간으로 보고 탐색할 수 있습니다. 이와 같은 시각화를 생성하려면 cuXFilter를 사용합니다:
nodes = tiles_xy_cdf
nodes['vertex']=nodes.index
nodes.columns=['x','y','vertex']
cux_df = fdf.load_graph((nodes, edge_df))
chart0 = cfc.graph(
edge_color_palette=['gray', 'black'],
timeout=200,
node_aggregate_fn='mean',
node_pixel_shade_type='linear',
edge_render_type='direct',#other option available -> 'curved', edge_transparency=0.5)
d = cux_df.dashboard([chart0], layout=clo.double_feature)
chart0.view()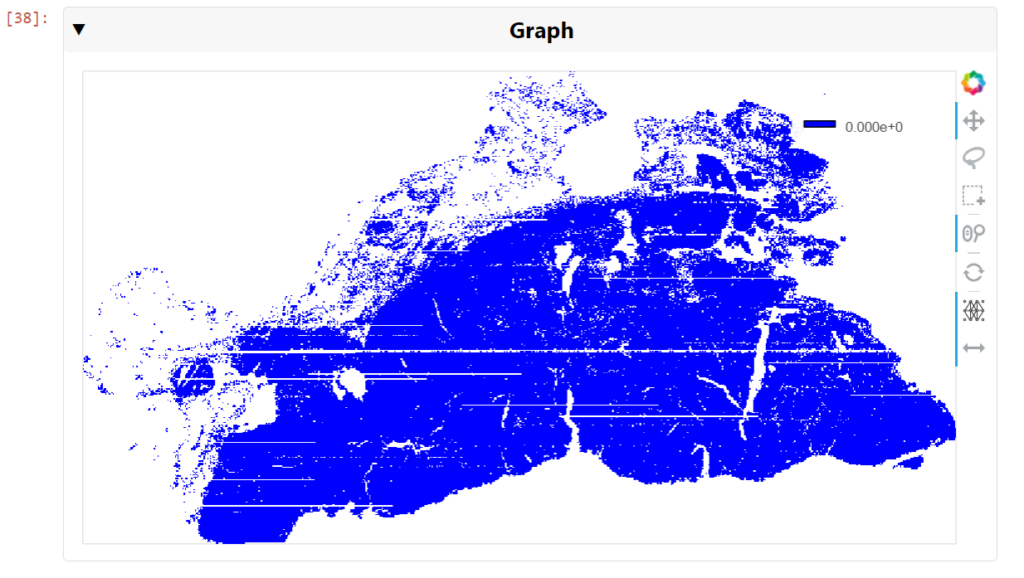
그런 다음 세포 핵 수준까지 이동 및 축소하여 가장 가까운 이웃의 클러스터를 볼 수 있습니다(그림 6).
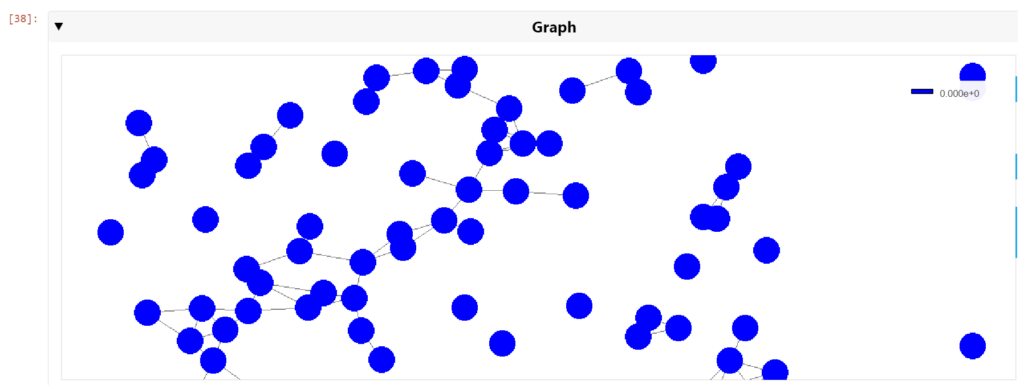
결론
원시 픽셀에서 인사이트를 도출하는 것은 어렵고 시간이 많이 소요될 수 있습니다. 몇 가지 강력한 도구와 기법을 대규모 이미지 문제에 적용하면 가장 까다로운 데이터도 거의 실시간으로 분석할 수 있습니다. ML 기능 외에도 RAPIDS와 같은 GPU 가속 도구는 DL 기반 방법이 생성하는 계산 기능을 해독하는 데 도움이 되는 강력한 시각화 기능도 제공합니다. 이 게시물에서는 DL, ML 그래프 및 GNN 방법을 사용하여 수집, 전처리, 추론, 후처리, 플로팅을 수행할 수 있는 엔드투엔드 도구 세트에 대해 설명했습니다.
RAPIDS와 MONAI를 시작하여 데이터에서 GPU의 성능을 최대한 활용하세요. 그리고 NVIDIA 개발자 포럼에서 MONAI 커뮤니티에 가입하세요.
관련 리소스
DLI 과정: RAPIDS cuDF로 데이터프레임 작업 속도 높이기
DLI 과정: 실시간 비디오 AI 애플리케이션 구축하기
DLI 과정: DeepStream을 사용한 지능형 비디오 분석을 위한 AI 워크플로우
GTC 세션: NVIDIA Holoscan*을 사용하여 고속 센서 AI 파이프라인 구축하기(2023년)
GTC 세션: CV-CUDA를 사용하여 AI 이미지 및 비디오 생성 툴을 최적화하는 런웨이(2023년)
웨비나: VPI 1.1을 사용하여 컴퓨터 비전 및 이미지 처리 가속화하기