
Fast and cost-effective whole genome sequencing and analysis can bring answers quickly to critically ill patients suffering from rare or undiagnosed diseases. Recent advances in accelerated clinical sequencing, such as the world-record-setting DNA sequencing technique for rapid diagnosis, are bringing us one step closer to same-day, whole-genome genetic diagnosis in a clinical setting.
A team led by Stanford University School of Medicine, NVIDIA, Google, UCSC, and Oxford Nanopore Technologies (ONT) recently used this technique to identify disease-associated genetic variants that resulted in a diagnosis in as little as 7 hours and 18 minutes, with results published in the NEJM and Nature Biotechnology.
This record-beating end-to-end genomic workflow is reliant on innovative technology and high-performance computation. It utilized long-read nanopore sequencing technology for better analysis of structural variants. This was implemented across 48 flow cells, with optimized methods enabling pore occupancy at 82%, rapidly generating 202 gigabases in just a couple of hours. Analysis of the output was distributed across a Google Cloud computing environment, including basecalling and alignment across 16 instances of 4 x V100 GPUs (64 GPUs total), and variant calling across 14 instances of 4 x P100 GPUs (56 GPUs total).
Since the January NEJM publication, the NVIDIA Clara team has been optimizing the same whole-genome workflow for the DGX-A100, giving clinicians and researchers the ability to deploy the same analysis as the world record approach on just eight A100 GPUs, and in just 4h10m for a 60x whole genome (Figure 1; benchmarked on the HG002 reference sample).
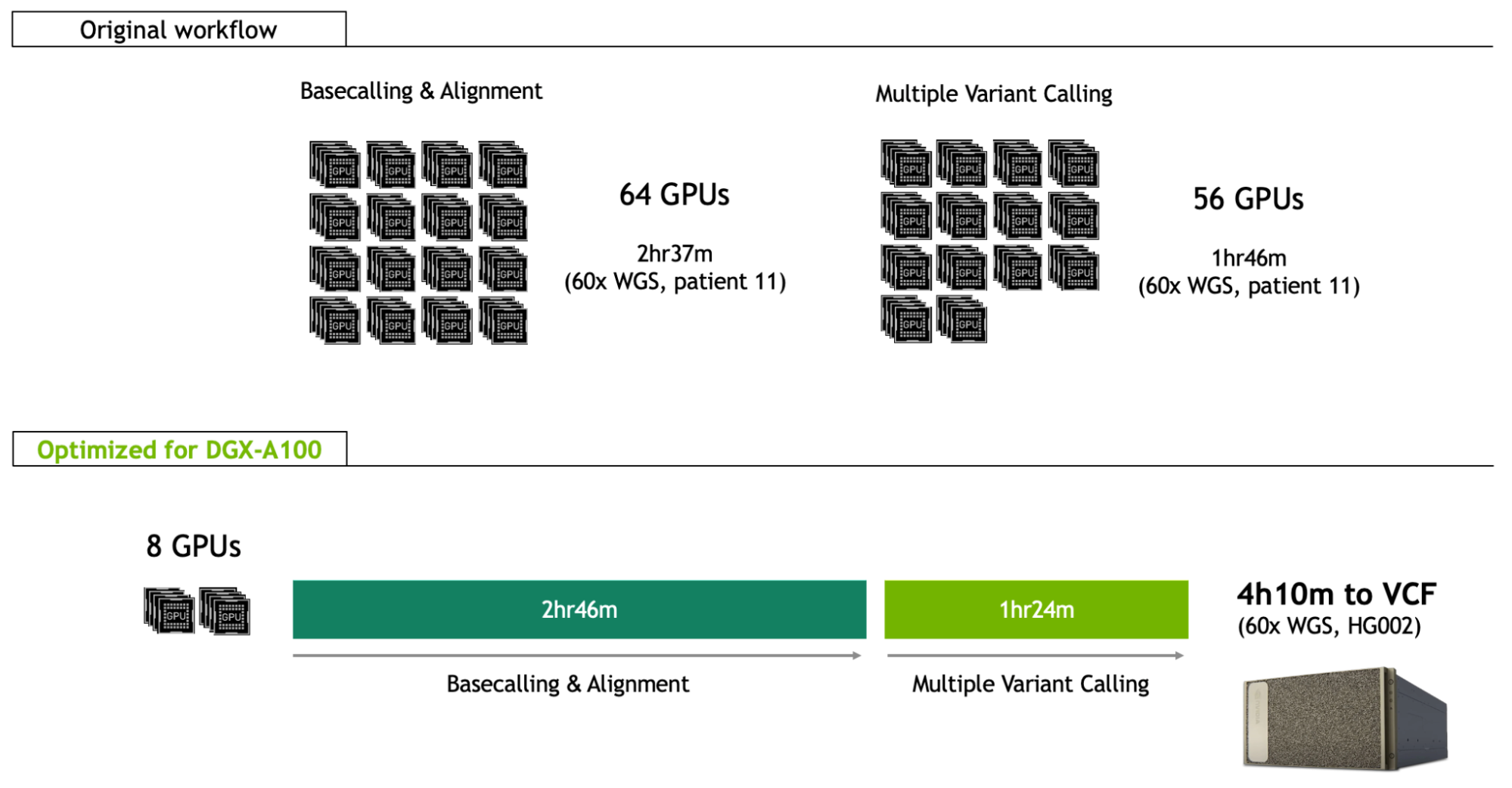
Not only does this enable fast analysis in a single server (8-GPU) framework that can be run locally, but it also lowers the cost per sample by two-thirds, from $568 to $183.
Basecalling and alignment
Basecalling is the process of classifying raw instrument signals into the bases A, C, G, and T of the genome. It is a computationally critical step in ensuring accuracy in all downstream analysis tasks. It is also an important data reduction step, reducing resultant data by approximately 10x.
At ~340 bytes per base, a single 60x coverage whole genome can easily be multiple terabytes in raw signals, as opposed to hundreds of gigabytes when processed. As such, it is beneficial for compute speed to rival sequencing output speed, which is non-trivial at a pace of ~450 bases per second through the 128,000 pores across 48 flow cells.
ONT’s PromethION P48 sequencer can generate as much as 10 terabases in a 72-hour run, equivalent to 96 human genomes (30x coverage).
The rapid classification task required for this already benefits from deep learning innovation and GPU acceleration. The core data processing toolkit for this purpose, Guppy, uses a recurrent neural network (RNN) for basecalling, with the option of three different architectures of smaller (faster) or larger (higher accuracy) recurrent layer sizes.
The main computational bottleneck in basecalling is the RNN kernel, which has benefited from GPU integration with ONT sequencers, such as the desktop GridION Mk1 that includes a single V100 GPU and the handheld MinION Mk1C that includes a Jetson Edge platform.
Alignment is the process of taking the resultant basecalled fragments of DNA, now in the form of character strings of As, Cs, Gs, and Ts, and determining the genome location where those fragments originated, assembling a full genome from the massively parallelized sequencing process. This essentially rebuilds the full length genome from many 100-100,000bp long reads. For the world-record-setting sample, this totaled around 13 million reads.
For the original world-record analysis, basecalling and alignment were run on separate instances of Guppy and Minimap2, respectively. In migrating this to a single-server DGX-A100 solution, and using Guppy’s integrated minimap2 aligner, you immediately save time on I/O and benefit from the tensor cores of the A100 for the RNN inferencing. Through balancing of basecalling and alignment across the DGX eight A100 GPUs and 256 CPU threads, respectively, the two processes can be overlapped perfectly to align reads concurrently with basecalling, resulting in no impact on total runtime (<1 min).
This brings the runtime of the basecalling and alignment step on the DGX-A100 to 2h 46m, which can also be overlapped with the sequencing itself. It is similar to the sequencing time expected for a 60x sample.
Variant calling
Variant calling is the portion of the workflow designed to identify all of the points in the newly assembled individual’s genome that differ from expected, compared to a reference genome. This involves scanning the full breadth of the genome to look for different types of variation. For example, this might include small single-base-pair variants all the way to large structural variants covering thousands of base-pairs. The world record pipeline used PEPPER-Margin-DeepVariant for small variants, and Sniffles for structural variants.
The PEPPER-Margin-DeepVariant approach is designed to optimize small variant calling for the long reads produced by nanopore sequencing.
- PEPPER identifies candidate variants through an RNN consisting of two bidirectional, gated, recurrent unit layers and a linear transformation layer.
- Margin then uses a hidden Markov model approach for a process called haplotyping, determining which variants have been inherited together from the maternal or paternal chromosomes. It passes this information to Google DeepVariant to use for maximum heterozygous variant calling accuracy.
- DeepVariant classifies final variants through a deep convolutional neural network, which is built on the Inception v2 architecture adapted specifically for DNA read pile-up input images.
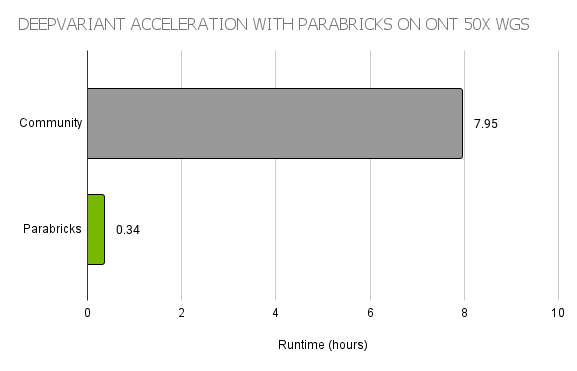
Overall, PEPPER-Margin-DeepVariant allows the faster neural network of PEPPER to scan the whole genome for candidates, and then uses the larger neural network of DeepVariant for high accuracy variant calling of those candidates. To accelerate this pipeline, the world-record workflow used Parabricks DeepVariant, a GPU-accelerated implementation providing >20x faster runtimes than the open-source version on CPU (Figure 2).
The Clara team took this acceleration further by modifying PEPPER-Margin to run in an integrated fashion, splitting the data by chromosome, and running the programs concurrently on GPU. PEPPER was also optimized for pipeline parameters such as batch sizes, number of workers, and number of callers, as well as upgrading PyTorch to enable support for NVIDIA Ampere Architecture acceleration of the RNN inference bottleneck.
For structural variant calling, Sniffles was upgraded to the recently released Sniffles 2, which is considerably more efficient, at 38x acceleration on CPU alone.
All these improvements put the runtime of the multiple variant calling stage at 1h 24m on the DGX-A100.
Powering real-time sequencing with NVIDIA DGX-A100
By optimizing the world-record DNA sequencing technique for DGX A100, the NVIDIA Clara team is powering real-time sequencing, simplifying a complex workflow on a single server, and cutting analysis costs by more than 50% while achieving state-of-the-art performance.
Learn more about deploying Parabricks with NVIDIA DGX A100.
Featured image courtesy of Oxford Nanopore Technologies