
Bioinformaticians are constantly looking for new tools that simplify and enhance genomic analysis pipelines. With over 60 tools, NVIDIA Clara Parabricks powers accurate and accelerated genomic analysis for germline and somatic workflows in research and clinical settings.
Today, we announce the release of NVIDIA Clara Parabricks 3.8, featuring:
- Rapid variant annotation tools.
- Tumor-only calling for clinical cancer workflows.
- Additional support on ampere GPUs.
The rapid increase in sequencing data demands faster variant call format (VCF) reading and writing speeds. Clara Parabricks 3.8 expands rapid variant annotation, post-variant calling, support of custom database annotation with snpswift, and variant consequence calling with bcftools. On Clara Parabricks, snpswift provides fast and accurate VCF database annotation and leverages a wide range of databases.
Advances in sequencing technologies are amplifying the role of genomics in clinical oncology. NVIDIA Clara Parabricks now provides tumor-only calling with somatic callers LoFreq and Mutect2 for clinical cancer workflows. Tumor-normal calling is available on Parabricks with LoFreq, Mutect2, Strelka2, SomaticSniper, Muse.
Genomic scientists can further accelerate genomic analysis workflows by running Clara Parabricks with a wider array of GPU architectures, including A6000, A100, A10, A30, A40, V100, and T4. This also supports customers using next-generation sequencing instruments powered by specific NVIDIA GPUs for basecalling that want to use the same GPUs for secondary analysis with Clara Parabricks.
Expanded rapid variant annotation
In the January release of Clara Parabricks 3.7, a new variant annotation tool was added that helps provide functional information of a variant for downstream genomic analysis. This is important, as correct variant annotation can assist with final conclusions of genomic studies and clinical diagnosis.
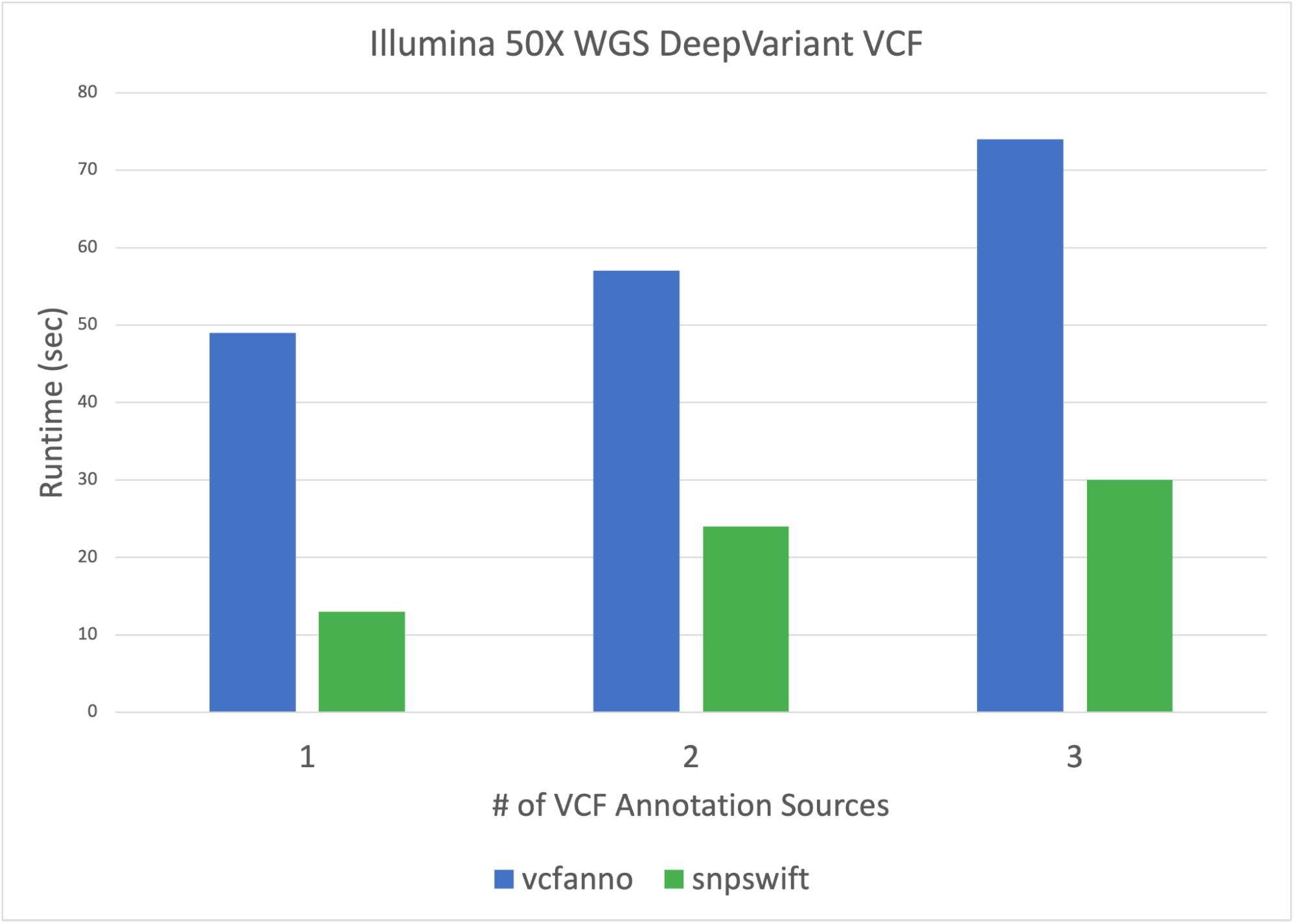
Parabricks’ variant annotation tool, snpswift, provides fast and accurate VCF database annotation that delivers results in shorter runtimes than other community variant annotation solutions such as vcfanno. Snpswift brings more functionality and acceleration while retaining the essential functionality of accurate allele-based database annotation of VCF files. The new snpswift tool also supports annotating a VCF file with gene name data from ENSEMBL, helping to make sense of coding variants.
Supported databases include dbSNP, gnomAD, COSMIC, ClinVar, and 1000 Genomes. Snpswift can annotate these jointly to provide important information for filtering VCF variants and interpreting their significance. Additionally, snpswift is able to annotate VCFs with information from an ENSEMBL GTF to add detailed information and leverage other widely used databases in the field.
In addition to these widely used databases, many research institutions and companies have their own rich internal datasets, which can provide valuable domain-specific information for each variant.
Snpswift in Clara Parabricks 3.8 now annotates VCFs with multiple custom TSV databases. Snpswift is also able to annotate a 6 million variant HG002 VCF with a custom TSV containing 500K variants in less than 30 seconds. Users are able to annotate VCFs with VCF, GTF, and TSV databases jointly—all with one command and in one run.
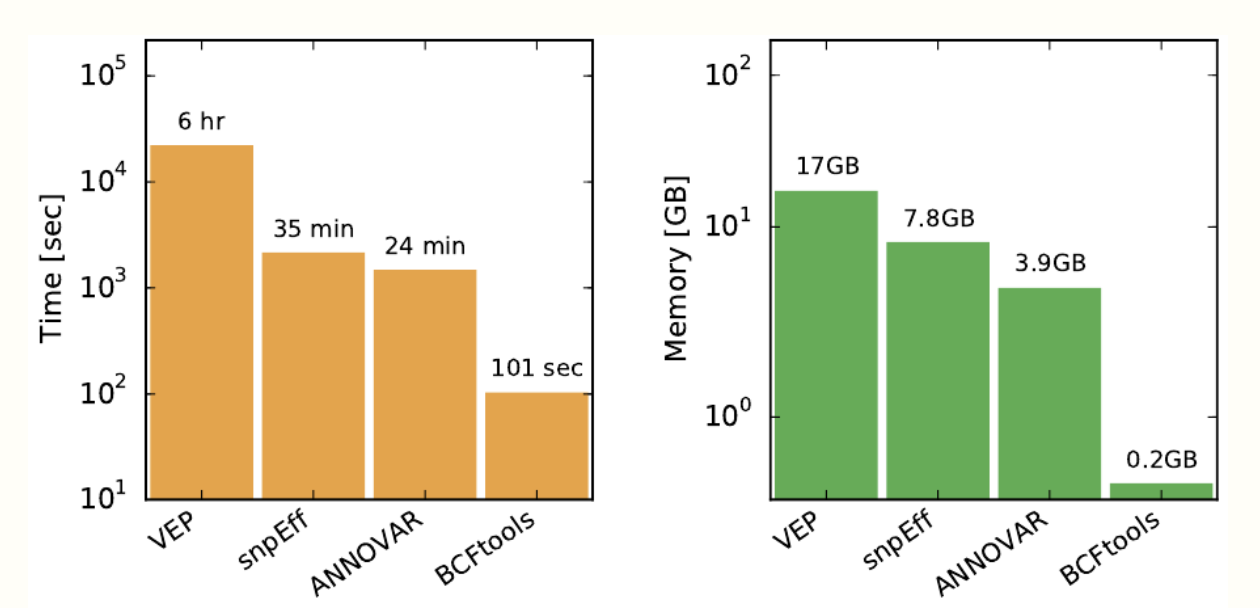
Finally, Clara Parabricks 3.8 has included consequence prediction. Predicting the functional outcome of a variant is a vital step in annotation for categorization and prioritization of genetic variants. Parabricks 3.8 offers a bcftoolscsq command that wraps the well known and extremely fast bcftools csq tool, providing haplotype-aware consequence predictions. This leads to phasing of variants in a VCF file to avoid common errors when variants affect the same codon.
Clara Parabricks has demonstrated 60x acceleration for state-of-the-art bioinformatics tools compared to CPU-based environments. End-to-end analysis of whole-genome workflows runs in 22 minutes and exome workflows in just 4 minutes. Large-scale sequencing projects and other whole-genome studies are able to analyze over 60 genomes a day on a single DGX server while reducing associated costs and generating more useful insights than ever before.
To get started on NVIDIA Clara Parabricks for your germline, cancer, and RNA-Seq analysis workflows, try a free 90-day trial. You can access Clara Parabricks on premise or in the cloud with AWS Marketplace.
An overview of how to annotate variants with Parabricks 3.8 and run consequence prediction using example data is available here as a GitHub Gist.
