
The release of NVIDIA Clara Parabricks v3.6 last summer added multiple accelerated somatic variant callers and novel tools for annotation and quality control of VCF files to the comprehensive toolkit for whole genome and whole exome sequencing analysis.
In the January 2022 release of Clara Parabricks v3.7, NVIDIA expanded the scope of the toolkit to new data types while continuing to improve upon existing tools:
- Added support for analysis of RNASeq analysis
- Added support for UMI-based gene panel analysis with an accelerated implementation of Fulcrum Genomics’
fgbiopipeline - Added support for
mutect2Panel of Normals (PON) filtering to bring the acceleratedmutectcallerin line with the GATK best practices for calling tumor-normal samples - Incorporated a
bam2fqmethod that enables accelerated realignment of reads to new references - Added support for short tandem repeat assays with
ExpansionHunter - Accelerated post-calling VCF analysis steps by up to 15X
- Updated
HaplotypeCallerto match GATK v4.1 and updatedDeepVariantto v1.1.
Clara Parabricks v3.7 significantly broadens the scope of what Clara Parabricks can do while continuing to invest in the field-leading whole genome and whole exome pipelines.
Enabling reference genome realignment with bam2fq and fq2bam
To address recent updates to the human reference genome and make realigning reads tractable for large studies, NVIDIA developed a new bam2fq tool. Parabricks bam2fq can extract reads in FASTQ format from BAM files, providing an accelerated replacement for tools like GATK SamToFastq or bazam.
Combined with Parabricks fq2bam, you can fully realign a 30X BAM file from one reference (for example, hg19) to an updated one (hg38 or CHM13) in 90 minutes using eight NVIDIA V100 GPUs. Internal benchmarks have shown that realigning to hg38 and rerunning variant calling captures several thousand more true-positive variants in the Genome in a Bottle HG002 truth set compared to relying solely on hg19.
The improvements in variant calls from realignment are almost the same as initially aligning to hg38. While this workflow was possible before, it was prohibitively slow. NVIDIA has finally made reference genome updates practical for even the largest of WGS studies in Clara Parabricks.
| ############# | |
| ## Download the 30X hg19-aligned bam from Google's public sequencing of HG002 | |
| ## and the respective BAI file. | |
| ############# | |
| wget https://storage.googleapis.com/brain-genomics-public/research/sequencing/grch37/bam/hiseqx/wgs_pcr_free/30x/HG002.hiseqx.pcr-free.30x.dedup.grch37.bam | |
| wget https://storage.googleapis.com/brain-genomics-public/research/sequencing/grch37/bam/hiseqx/wgs_pcr_free/30x/HG002.hiseqx.pcr-free.30x.dedup.grch37.bam.bai | |
| ############# | |
| ## Prepare the references so we can realign reads | |
| ############# | |
| ## Download the original hg19 / hsd37d5 reference | |
| ## and create and FAI index | |
| wget ftp://ftp-trace.ncbi.nih.gov/1000genomes/ftp/technical/reference/phase2_reference_assembly_sequence/hs37d5.fa.gz | |
| gunzip hs37d5.fa.gz | |
| samtools faidx hs37d5.fa | |
| ## Download GRCh38 | |
| wget ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/000/001/405/GCA_000001405.15_GRCh38/seqs_for_alignment_pipelines.ucsc_ids/GCA_000001405.15_GRCh38_no_alt_analysis_set.fna.gz | |
| gunzip GCA_000001405.15_GRCh38_no_alt_analysis_set.fna.gz | |
| ## Make a .fai index using samtools faidx | |
| samtools faidx GCA_000001405.15_GRCh38_no_alt_analysis_set.fna | |
| ## Create the BWA indices | |
| bwa index GCA_000001405.15_GRCh38_no_alt_analysis_set.fna | |
| ## Download the Gold Standard indels from 1kg to use as your known-sites file. | |
| wget https://storage.googleapis.com/genomics-public-data/resources/broad/hg38/v0/Mills_and_1000G_gold_standard.indels.hg38.vcf.gz | |
| ## Also grab the tabix index for the file | |
| wget https://storage.googleapis.com/genomics-public-data/resources/broad/hg38/v0/Mills_and_1000G_gold_standard.indels.hg38.vcf.gz.tbi | |
| ############ | |
| ## Run the bam2fq tool to extract reads from the BAM file | |
| ## Adjust the --num-threads argument to reflect the number of cores on your system. | |
| ## With 8 GPUs and 64 vCPUs this should take ~45 minutes. | |
| ############ | |
| time pbrun bam2fq \ | |
| --ref hs37d5.fa \ | |
| --in-bam HG002.hiseqx.pcr-free.30x.dedup.grch37.bam \ | |
| --out-prefix HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq \ | |
| --num-threads 64 | |
| ############## | |
| ## Run the fq2bam tool to align reads to GRCh38 | |
| ############## | |
| time pbrun fq2bam \ | |
| --in-fq HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq_1.fastq.gz HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq_1.fastq.gz \ | |
| --ref Homo_sapiens_assembly38.fasta \ | |
| --knownSites Mills_and_1000G_gold_standard.indels.hg38.vcf.gz \ | |
| --out-bam HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq.hg38.bam \ | |
| --out-recal-file HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq.hg38.BQSR-REPORT.txt |
More options for RNASeq transcript quantification and fusion calling in Clara Parabricks
With version 3.7, Clara Parabricks adds two new tools for RNASeq analysis as well.
Transcript quantification is one of the most performed analyses for RNASeq data. Kallisto is a rapid method for expression quantification that relies on pseudoalignment. While Clara Parabricks already included STAR for RNASeq alignment, Kallisto adds a complementary method that can run even faster.
Fusion calling is another common RNASeq analysis. In Clara Parabricks 3.7, Arriba provides a second method for calling gene fusions based on the output of the STAR aligner. Arriba can call significantly more types of events than STAR-Fusion, including the following:
- Viral integration sites
- Internal tandem duplications
- Whole exon duplications
- Circular RNAs
- Enhancer-hijacking events involving immunoglobulin and T-cell receptor loci
- Breakpoints in introns and intergenic regions
Together, the addition of Kallisto and Arriba make Clara Parabricks a comprehensive toolkit for many transcriptome analyses.
Simplifying and accelerating gene panel and UMI analysis
While whole genome and whole exome sequencing are increasingly common in both research and clinical practice, gene panels dominate the clinical space.
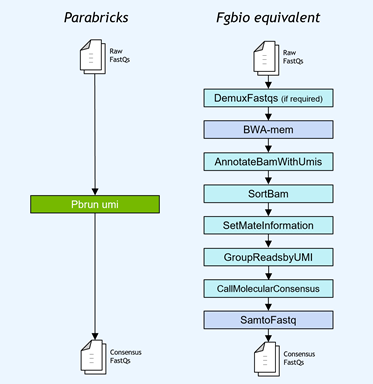
Gene panel workflows commonly use unique molecular identifiers (UMIs) attached to reads to improve the limits of detection for low-frequency mutations. NVIDIA accelerated the Fulcrum Genomics fgbio UMI pipeline and consolidated the eight-step pipeline into a single command in v3.7, with support for multiple UMI formats.
Detecting changes in short tandem repeats with ExpansionHunter
Short tandem repeats (STRs) are well-established causes of certain neurological disorders as well as historically important markers for fingerprinting samples for forensic and population genetic purposes.
NVIDIA enabled genotyping of these sites in Clara Parabricks by adding support for ExpansionHunter in version 3.7. It’s now easy to go from raw reads to genotyped STRs entirely using the Clara Parabricks command-line interface.
Improving MuTect somatic mutation calls with PON support
It is common practice to filter somatic mutation calls against a set of mutations from known normal samples, also called a Panel of Normals (PON). NVIDIA added support for both publicly available PON sets and custom PONs to the mutectcaller tool, which now provides an accelerated version of the GATK best practices for somatic mutation calling.
Accelerating post-calling VCF annotation and quality control
In the v3.6 release, NVIDIA added the vbvm, vcfanno, frequencyfiltration, vcfqc, and vcfqcbybam tools that made post-calling VCF merging, annotation, filtering,filtering, and quality control easier to use.
The v3.7 release improved upon these tools by completely rewriting the backend of vbvm, vcfqc and vcfqcbybam, all of which are now more robust and up to 15x faster.
In the case of vcfanno, NVIDIA developed a new annotation tool called snpswift, which brings more functionality and acceleration while retaining the essential functionality of accurate allele-based database annotation of VCF files. The new snpswift tool also supports annotating a VCF file with gene name data from ENSEMBL, helping to make sense of coding variants. While the new post-calling pipeline looks similar to the one from v3.6, you should find that your analysis runs even faster.
| #!/bin/bash | |
| ######################## | |
| ## In this gist, we'll reuse the commands from our 3.6 tutorial to align reads and generate BAM files. | |
| ## Check out the full post at https://medium.com/@johnnyisraeli/accelerating-germline-and-somatic-genomic-analysis-of-whole-genomes-and-exomes-with-nvidia-clara-e3deeae2acc9 | |
| and Gists at: | |
| ## https://gist.github.com/edawson/e84b2785db75d3c0aea9cc6a59969d45#file-full_pipeline_and_data_prep_parabricks3-6-sh | |
| ## and | |
| ## https://gist.github.com/edawson/e84b2785db75d3c0aea9cc6a59969d45#file-step_1_align_reads_parabricks3-6-sh | |
| ########### | |
| ########### | |
| ## We'll run this tutorial on a GCP VM with 64 vCPUs, 240GB of RAM, and 8x NVIDIA V100 GPUs | |
| ## To save costs, you can also run this on a GCP VM with 32 vCPUS, 120GB of RAM, and 4x V100 GPUs | |
| ########### | |
| ## After aligning our reads, we'll rerun the variant calling stages of our past gist | |
| ## since we've updated the haplotypecaller and DeepVariant tools. We'll | |
| ## also run Strelka2 as an additional variant caller. | |
| ## | |
| ## After that, we'll merge our VCFs to generate a union callset and an intersection VCF | |
| ## with variants called by all three variant callers, annotate our new intersection VCF, | |
| ## and remove variants that fail certain criteria for population frequency. | |
| ## Finally, we'll run our vcfqc and vcfqcbybam tools to generate simple quality control reports. | |
| ############# | |
| ################ | |
| ## HaplotypeCaller | |
| ## This step should take roughly 15 minutes on our 8xV100 VM. | |
| ################ | |
| time pbrun haplotypecaller \ | |
| --ref ~/refs/Homo_sapiens_assembly38.fasta \ | |
| --in-bam HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq.hg38.bam \ | |
| --in-recal-file HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq.hg38.BQSR-REPORT.txt \ | |
| --out-variants HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq.hg38.pb.haplotypecaller.vcf | |
| ################ | |
| ## DeepVariant | |
| ## This step should take approximately 20 minutes on an 8xV100 VM | |
| ################ | |
| time pbrun deepvariant \ | |
| --ref Homo_sapiens_assembly38.fasta \ | |
| --in-bam HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq.hg38.bam \ | |
| --out-variants HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq.hg38.pb.deepvariant.vcf | |
| ############### | |
| ## Strelka | |
| ## This step should take ~10 minutes on a 64-core VM. | |
| ############### | |
| time pbrun strelka \ | |
| --ref Homo_sapiens_assembly38.fasta \ | |
| --in-bams HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq.hg38.bam \ | |
| --out-prefix HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq.hg38.pb.strelka \ | |
| --num-threads 64 | |
| ## Copy strelka results to current directory. | |
| cp HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq.hg38.pb.strelka.strelka_work/results/variants/variants.vcf.gz* . | |
| ## BGZIP and tabix-index the deepvariant VCFs | |
| bgzip -@16 HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq.hg38.pb.deepvariant.vcf | |
| tabix HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq.hg38.pb.deepvariant.vcf.gz | |
| ## BGZIP and tabix index the haplotypecaller VCFs | |
| bgzip -@16 HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq.hg38.pb.haplotypecaller.vcf | |
| tabix HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq.hg38.pb.haplotypecaller.vcf.gz | |
| ## Run the votebasedvcfmerger tool to generate a union and intersection VCF. | |
| time pbrun votebasedvcfmerger \ | |
| --in-vcf strelka:variants.vcf.gz \ | |
| --in-vcf deepvariant:HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq.hg38.pb.deepvariant.vcf.gz \ | |
| --in-vcf haplotypecaller:HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq.hg38.pb.haplotypecaller.vcf.gz \ | |
| --min-votes 3 | |
| --out-dir HG002.realign.vbvm | |
| ## The HG002.realign.vbvm directory should now contain a | |
| ## unionVCF.vcf file with the union callset of HaplotypeCaller, Strelka, and DeepVariant | |
| ## and aa filteredVCF.vcf file with only calls produced by all three callers. | |
| ## Annotate the intersection VCF with gnomAD, ClinVar, 1000 Genomes | |
| ## Download our annotation VCFs and tabix indices | |
| wget https://ftp.ncbi.nlm.nih.gov/pub/clinvar/vcf_GRCh38/clinvar.vcf.gz | |
| wget https://ftp.ncbi.nlm.nih.gov/pub/clinvar/vcf_GRCh38/clinvar.vcf.gz.tbi | |
| wget https://storage.googleapis.com/gcp-public-data--gnomad/release/2.1.1/liftover_grch38/vcf/exomes/gnomad.exomes.r2.1.1.sites.liftover_grch38.vcf.bgz | |
| wget https://storage.googleapis.com/gcp-public-data--gnomad/release/2.1.1/liftover_grch38/vcf/exomes/gnomad.exomes.r2.1.1.sites.liftover_grch38.vcf.bgz.tbi | |
| wget http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/1000_genomes_project/release/20181203_biallelic_SNV/ALL.wgs.shapeit2_integrated_v1a.GRCh38.20181129.sites.vcf.gz | |
| wget http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/1000_genomes_project/release/20181203_biallelic_SNV/ALL.wgs.shapeit2_integrated_v1a.GRCh38.20181129.sites.vcf.gz.tbi | |
| ## Download an Ensembl GTF to annotate the VCF file with gene names | |
| wget http://ftp.ensembl.org/pub/release-105/gtf/homo_sapiens/Homo_sapiens.GRCh38.105.gtf.gz | |
| ## Unzip the GTF file and add the "chr" prefix to the chromosome names (Ensembl excludes this prefix by default. | |
| gunzip Homo_sapiens.GRCh38.105.gtf.gz | |
| awk '{if($0 !~ /^#/) print "chr"$0; else print $0}' Homo_sapiens.GRCh38.105.gtf > Homo_sapiens.GRCh38.105.chr.gtf | |
| time pbrun snpswift \ | |
| --input-vcf HG002.realign.vbvm/filteredVCF.vcf \ | |
| --anno-vcf 1000Genomes:ALL.wgs.shapeit2_integrated_v1a.GRCh38.20181129.sites.vcf.gz \ | |
| --anno-vcf gnomad_v2.1.1:gnomad.exomes.r2.1.1.sites.liftover_grch38.vcf.bgz \ | |
| --anno-vcf ClinVar:clinvar.vcf.gz \ | |
| --ensembl Homo_sapiens.GRCh38.105.chr.gtf \ | |
| --output-vcf HG002.realign.3callers.annotated.vcf | |
| ################## | |
| ## frequencyfiltration | |
| ## Next we'll filter our VCF to remove variants with 1000Genomes allele frequency > 0.05 | |
| ## and gnomAD AF < 0.05 | |
| ################## | |
| time pbrun frequencyfiltration \ | |
| --in-vcf HG002.realign.3callers.annotated.vcf \ | |
| --and-expression "1000Genomes_AF < 0.05" \ | |
| --and-expression "gnomad_v2.1.1_AF < 0.05" \ | |
| --out-vcf HG002.realign.3callers.annotated.filtered.vcf | |
| ################## | |
| ## Finally, we'll run our automated vcfqc tool to generate some | |
| ## basic QC stats. The vcfqcbybam tool could also be run | |
| ## to produce QC stats using an auxilliary BAM file (e.g., when variant calls don't have the desired fields). | |
| ################## | |
| time pbrun vcfqc --in-vcf HG002.realign.3callers.annotated.filtered.vcf \ | |
| --output-dir HG002.realign.3callers.annotated.filtered.qc \ | |
| --depth haplotypecaller_DP --allele-depth deepvariant_AD |
Summary
With Clara Parabricks v3.7, NVIDIA is demonstrating a commitment to making Parabricks the most comprehensive solution for accelerated analysis of genomic data. It is an extensive toolkit for WGS, WES, and now RNASeq analysis, as well as gene panel and UMI data.
For more information about version 3.7, see the following resources:
- Clara Parabricks 3.7 Brings Optimized and Accelerated Workflows for Gene Panels
- Clara Parabricks documentation
Try out Clara Parabricks for free for 90 days and run this tutorial on your own data.
