Molecular dynamics simulations and AI-powered computational chemistry are playing a key role in the fight against COVID-19, providing atomic-scale insights to viral mechanisms including virus-to-cell fusion, viral protein function, and ultimately possible therapeutics.
Molecular dynamics simulations offer accurate approximations of real molecular behavior providing a tool to better understand how drugs might bind to a protein. Applications like OPENMM, NAMD, and GROMACS are well-suited for GPUs because they lend themselves to data-parallel implementations.
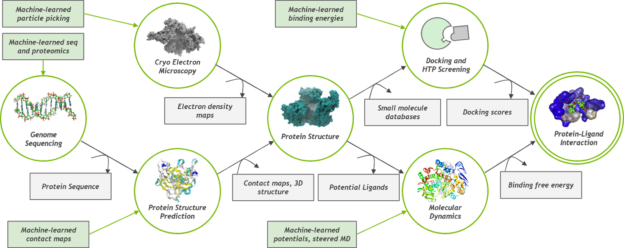
Simulations in these applications can run for several hundred nanoseconds. The ability to run longer simulations in less time is of extreme importance because it can show that the drug was able to modify the behavior of the protein without causing any side effects. For example, events such as folding and docking can take micro- and milliseconds to occur. A simulation that gets cut short because of limited compute power may miss these important aspects of protein and compound simulations.
Watch this webinar, part of the Compute4COVID technical series, to learn more about AI and machine learning speeding up molecular simulation via machine-learned force-fields, enhanced free energy methods, and generative methods that complement drug screening pipelines.
More Information
Discover how AI, accelerated computing, and technology are contributing to the worldwide battle against the novel coronavirus in our COVID-19 Research Hub.
