
Dr. Avantika Lal is a deep learning and genomics scientist at NVIDIA and was previously a researcher at Stanford University. She holds a PhD in genomics and is an expert in the genomics of infectious diseases and cancer. At NVIDIA, she develops artificial intelligence techniques to analyze genomic data, and applies these methods to understand human biology and develop new and targeted treatments for disease.
Despite the many improvements in treating COVID-19 after the beginning of the pandemic, it is still difficult to treat. Part of that is due to gaps in our understanding of the basic biology of how the virus affects human cells. Developing effective treatments is dependent on a better understanding of the fundamental mechanisms of this disease.
Published today in Nature Communications Biology, the research of NVIDIA’s Avantika Lal has unlocked some key findings about these fundamental mechanisms, discovering genes, proteins, and biological processes in human cells that are specifically altered from SARS-CoV-2. You can view the full research article here.

What inspired you to enter the field of AI/DL? How did you first fall in love with the field?
During my PhD in genetics, I realized that genetics was quickly becoming a science of big data analytics. It became increasingly clear that the future of biology lay in mining massive genomic datasets to understand the relationships between biological components and being able to make new predictions based on that. AI and deep learning were the most promising tools to achieve this. In the years after that, I’ve enjoyed being part of the change as these methods become more and more common in biology and produce insights that wouldn’t have been possible otherwise.
Can you talk a bit about your current research?
I’ve been working with a team of researchers from eight different institutions to study the genetic response of human cells to COVID-19 infection by mining public datasets. When cells are infected by a virus, it triggers an immune response that changes the activity of the cell and signals to the immune system. The virus also ‘hijacks’ many components of the cell to support its lifecycle, which disrupts their normal functioning.
We analyzed public data from DNA and RNA sequencing experiments to decipher and in some cases to predict, which genes, proteins, and biological processes are affected in human lung cells infected by the SARS-CoV-2 virus. We see indications that cells infected by SARS-CoV-2 show changes that are not common in infections with other respiratory viruses. For example, we identified 64 genes whose activity is consistently changed across multiple types of human lung cells infected by the SARS-CoV-2 virus but remain unaffected by other viruses.
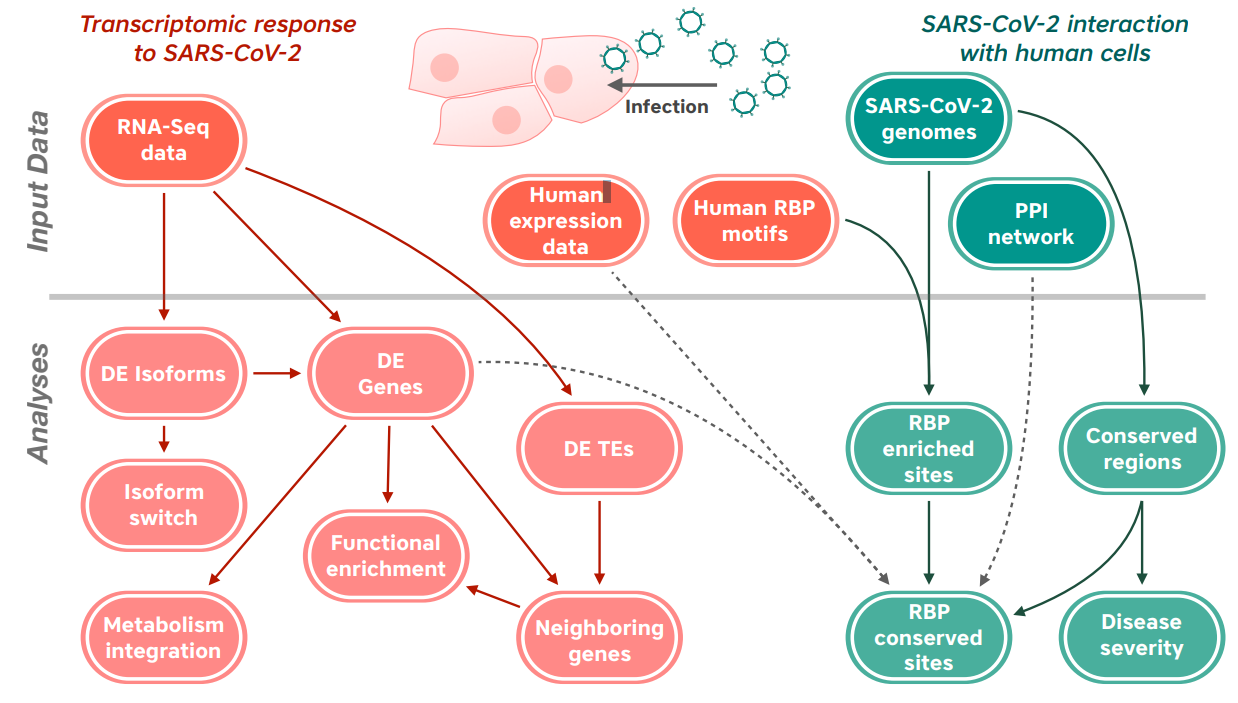
What motivated you to tackle the challenges of COVID-19?
Despite all that we’ve learned from the pandemic’s beginnings, it is still difficult to treat COVID-19. That’s partly because we don’t have the full picture of how the virus affects human cells. Understanding the fundamental mechanisms of this disease will help us develop effective treatments.
What are the biggest research challenges that you faced in this project?
Data availability was a challenge early in the pandemic, when few experimental datasets on SARS-CoV-2 infected cells were publicly available. New data has now come out and it’s great to see that most scientists are making COVID-19 data available before journal publication.
Another challenge is that computational models used to predict genetic interactions are still far from perfect and frequently produce false positive results. Ideally, these methods can be used to filter out interesting candidates that can be validated experimentally. We are excited to see experimental studies emerging to help us assess the accuracy of our computational predictions.
What NVIDIA technologies are you using to overcome these challenges?
One of our analyses aims to predict interactions between human RNA-binding proteins and the RNA genome of SARS-CoV-2. Such proteins bind to RNA molecules depending on the sequence of the RNA, and could affect the replication, function, and stability of SARS-CoV-2. The probability of a protein binding to an RNA sequence can be encoded as a matrix representation called a position-weight matrix.
We convolved the RNA genome sequence of the virus with these matrices to identify potential sites where the protein may bind to RNA. We are currently accelerating this operation by using TensorFlow on GPUs. In addition, deep learning methods have emerged in the last few years that are more accurate at predicting RNA-protein interactions. By combining deep learning and conventional predictions on GPUs, we aim to develop a framework to predict RNA-protein interactions quickly and accurately in SARS-CoV-2 and other viral genomes.
What is the impact of your research on the larger COVID-19 research community?
Our computational analysis discovered genes, proteins, and biological processes in human cells that are specifically altered in SARS-CoV-2 infection. We hope that this understanding may translate into better therapeutics to treat COVID-19. In many cases, the changes that we see appear to be specific to SARS-CoV-2 and are not observed in other respiratory viral infections. This helps us understand the unique properties of COVID-19.
What’s the next evolution of your research? Where are you hoping that this leads?
Currently, we aim to extend some of our analyses to other viral infections, in addition to COVID-19. This knowledge base will improve scientific understanding of viral infections in general, and further our understanding of COVID-19 by enabling more thorough comparisons to other viruses.
What unique opportunities has COVID-19 brought to the global research community?
Our study is a great example of how COVID-19 has inspired collaboration across borders in the research community. This study was performed by 13 authors from eight institutions in six countries. I’m very happy to see the commitment of the scientific community to openly sharing COVID-19 relevant data without waiting for publication, as well as the initiative taken by several journals to make all COVID-19 related papers available without a paywall.
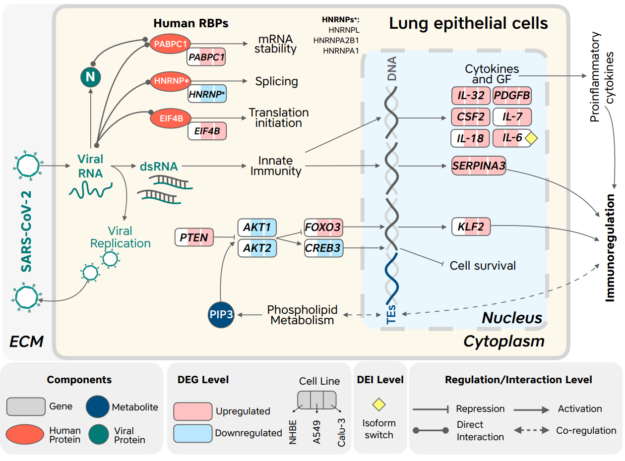
Figure 2 shows that the factors specific to SARS-CoV-2 infection include human RBPs whose binding sites are enriched and conserved in the SARS-CoV-2 genome but not in the genomes of related viruses; and genes, isoforms and metabolites that are consistently altered in response to SARS-CoV-2 infection of lung epithelial cells but not in infection with the other tested viruses; ECM (extracellular matrix).
In terms of technological advances to further your research area, what are you looking forward to next?
We’ve seen the widespread application of machine learning and deep learning techniques in medical imaging to diagnose COVID-19. Machine learning in genomics is still a developing field, and there are few studies that use these capabilities to analyze COVID-19 genomic data. With more and larger datasets becoming available, I hope to see more applications of ML methods to extract biological insights from COVID-19 genomic data.
How has COVID-19 shaped–or reshaped–your research workflow? How will the research community adapt to the ‘new normal?’
Being a computational biologist, I’m fortunate in being able to continue my research relatively normally. The biggest disruption has been the cancellation or virtualization of major conferences where researchers would normally connect and discuss their ongoing work. As a community, we must come up with better ways for researchers to meet, connect, collaborate, and seek advice over virtual platforms.
What advice would you give to the next generation of researchers?
Research is becoming more and more interdisciplinary–it’s good to read widely about the latest developments in other domains and think about how they could be applied to your field. AI in genomics is a good example of this!
More reading
To learn more about Avantika’s work in genomic analysis, read her latest post, Accelerating Single Cell Genomic Analysis using RAPIDS.
