
NAMD, a widely used parallel molecular dynamics simulation engine, was one of the first CUDA-accelerated applications. Throughout the early evolution of CUDA support in NAMD, NVIDIA GPUs and CUDA grew tremendously in both performance and capabilities. For more information, see Accelerating Molecular Modeling Applications with Graphics Processors and Adapting a Message-driven Parallel Application to GPU-Accelerated Clusters.
The arrival of the Volta GPU generation brought NAMD to bear on record-setting simulation sizes on the Oak Ridge National Laboratory (ORNL) Summit system. For more information, see Early Experiences Porting the NAMD and VMD Molecular Simulation and Analysis Software to GPU-Accelerated OpenPOWER Platforms and Scalable Molecular Dynamics with NAMD on the Summit System.
However, because host CPUs and PCIe had not matched the performance gains made by GPUs during the same period, the original NAMD software design became a barrier to further performance gains for small- and moderate-sized molecular dynamics simulations. The performance provided by Volta GPUs effectively made NAMD v2 CPU-bound and eliminating this performance limitation required a different approach.
Projecting expected GPU performance gains forward and anticipating the increasing availability of a variety of dense-GPU HPC platforms, the NAMD team embarked on a significant shift in strategy away from the traditional “GPU-accelerated” scheme, toward a completely “GPU-resident” mode of operation for NAMD v3.
The new GPU-resident mode of NAMD v3 targets single-node single-GPU simulations, and so-called multi-copy and replica-exchange molecular dynamics simulations on GPU clusters, and dense multi-GPU systems like the DGX-2 and DGX-A100. The NAMD v3 GPU-resident single-node computing approach has greatly reduced the NAMD dependence on CPU and PCIe performance, leading to tremendous performance for small and moderate-sized simulations on state-of-the-art NVIDIA Ampere GPUs and dense multi-GPU platforms such as DGX-A100.
The early results presented in this post are very exciting, but we must note that more work remains to be done. We expect performance to grow further as NAMD v3 matures and we later begin work on strong scaling with NVLink– and NVSwitch-connected systems.
NAMD v3 becomes GPU-bound after switching to the “GPU-resident” mode. This makes v3 up to 1.9X faster than v2.13 on the same GPU. Being GPU-bound also makes v3 benefit more from using newer GPUs: v3 is up to 1.4X faster on the new A100 than on V100. Finally, being GPU-bound makes v3 scale much better for multi-copy simulation on multi-GPU systems. As the combined results of those software and hardware progress, on the 8-GPU DGX systems, you see that v3 throughput on A100 is up to 9X higher than v2.13 on V100.
NAMD v3 performance optimization
NAMD employs a time-stepping algorithm to propagate molecular systems in time. Each timestep is usually 1 or 2 femtoseconds long, and the phenomena to observe usually happens on the nanosecond to microsecond scale. It’s necessary to perform millions and millions of timesteps.
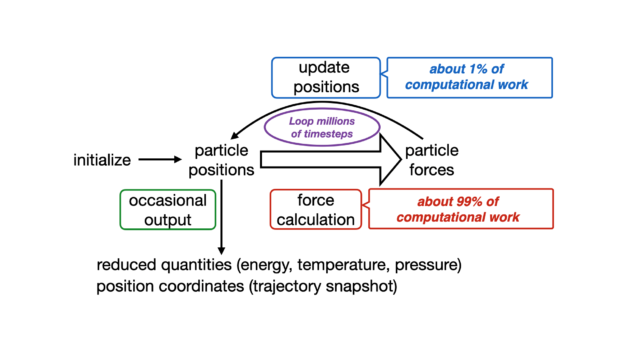
The conventional timestep is usually comprised of four main computational bottlenecks:
- Short-range nonbonded forces: Evaluation of force and energy terms of atoms are inside a specified cutoff and considered to interact directly ≈ 90% overall FLOPS.
- Long-range forces through Particle-mesh Ewald (PME): Fast approximation of long-range electrostatic interactions are on the reciprocal space, which involves a few Fourier transforms ≈ 5% overall FLOPS.
- Bonded forces: Bonded terms coming from a molecular topology are calculated, weighted, and applied to their corresponding atoms ≈ 4% overall FLOPS.
- Numerical integration: Calculated forces are applied to atom velocities and the system is propagated in time through a velocity-verlet algorithm ≈ 1% overall FLOPS.
As one of the earliest scientific codes to adopt GPU acceleration, the most recent releases of NAMD (2.13 and 2.14) can offload all force terms and fetch the results back to the CPU host, where numerical integration takes place. This scheme allowed NAMD to use all machine resources efficiently, and it was able to extract full performance of Maxwell and older GPU generations. However, after the Pascal GPU architecture was released, NAMD has been having trouble fully occupying GPUs with work.
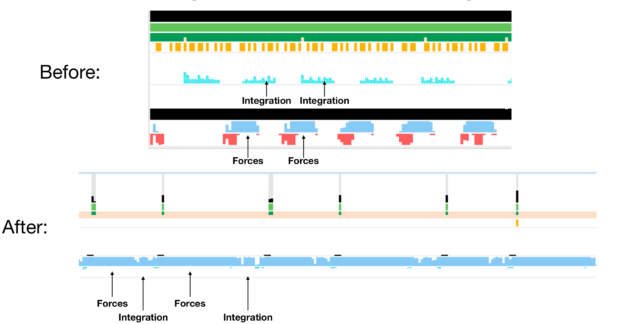
To help you understand what is really going on, we used NVIDIA Nsight Systems to generate a timeline of NAMD execution and track the GPU activity across the simulation.

As you track the GPU activity, gaps in the blue strip reveal that the GPU is idling across the simulation due to the remaining task happening on the CPU: numerical integration. The takeaway message is that modern GPUs are so efficient that delegating even so small a part as 1% of the overall FLOPs to the CPU is responsible for bottlenecking the entire simulation. To keep benefiting from GPU processing power, move the numerical integration step to GPUs as well.
To achieve scalable parallelism, NAMD spatially decomposes the molecular system into a series of sub-domains, much like a three-dimensional patchwork quilt where each patch represents a subset of atoms. Normally, patches themselves are responsible for storing the relevant atomic information, such as total forces, positions, and velocities, and to propagate their subset of atoms in time using the velocity-verlet algorithm.
Moving this code to the GPU involves handling how patches are organized. Blindly porting numerical integration operations to CUDA is simple enough as most of its algorithms are data-parallel. However, due to the focus on scalability, patches are too fine-grained to fully occupy GPUs with enough integration operations. Moreover, to deal with remote forces that might arrive from different computational nodes, NAMD contains a default infrastructure to communicate GPU-calculated forces to patches allocated on remote nodes.
So, to benefit from the GPU power during integration, you can’t rely on the existing NAMD patch-wise integration scheme. However, due to remote forces arriving from different nodes, you must go through that infrastructure, as information needs to be communicated.
To deal with this problem, we decided it would be fruitful to have a special code path to handle single-node simulations, so that we can avoid having to go through the CPU mechanisms for communicating forces. We developed a scheme to do the following:
- Fetch forces as soon as they are calculated from the GPU force kernels.
- Format them into a structure-of-arrays data structure to allow for a regular memory access pattern during integration.
- Aggregate all data of all patches in the simulation to launch a single integration task (with multiple kernels) for the entire molecular system.
Patches no longer hold relevant atomic information and are just there to represent the spatial decomposition itself.

With this scheme in place, it is possible to use the CPU for issuing kernels and handling I/O only, while using modern GPUs fully to perform the relevant math.
As we said earlier, most integration kernels are data-parallel operations. However, some of them require caution while porting. Usually, if the user requires longer timesteps (2 femtoseconds or more), it is recommended to constraint the hydrogen bond lengths during the simulation (usually called rigid bonds constraints).
This operation runs during numerical integration and could become a bottleneck if done incorrectly. We have devised a single kernel to solve all constraints of the system, adapting the SETTLE algorithm to handle waters and the Matrix-SHAKE variant for other hydrogen bonds that do not belong to waters.
This scheme is available on alpha builds of the NAMD3.0 binary and currently only supports single-GPU runs. Upcoming support for single-trajectory simulations using multiple, fully interconnected NVIDIA GPUs, is currently being developed.
Getting good performance with NAMD v3
To enable the fast, single-GPU code path, add the following option to the NAMD config command:
--with-single-node-cuda
In your NAMD configuration file, set CUDASOAintegrate to on.
For good performance, it is important to set the following performance tuning parameters properly:
stepsPerCycle(NAMD 2.x default = 20)pairlistsPerCycle(NAMD 2.x default = 2)margin(NAMD 2.x = 0)
If these are left as UNDEFINED in the simulation configuration file with CUDASOAintegrate enabled, NAMD automatically sets them to the recommended values as follows:
stepsPerCycle 400pairlistsPerCycle 40margin 8
In addition, frequent output limits performance. Most importantly, outputEnergies (defaulting in NAMD 2.x to 1) should be set much higher. If left as UNDEFINED in the simulation configuration file with CUDASOAintegrate enabled, NAMD automatically sets the following parameters to the same value as stepsPerCycle, again with this default:
outputEnergies 400outputTimings 400
The fast path doesn’t support minimization yet. For inputs that use minimization, minimization and dynamics should be performed in two separate NAMD runs. The CUDASOAintegrate parameter should be set to off for the minimization run. Then the dynamics run can be restarted with the fast path.
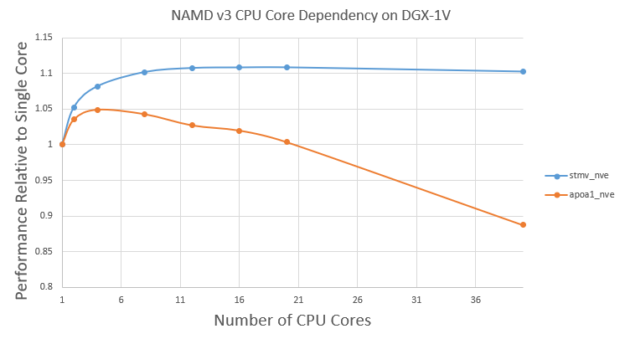
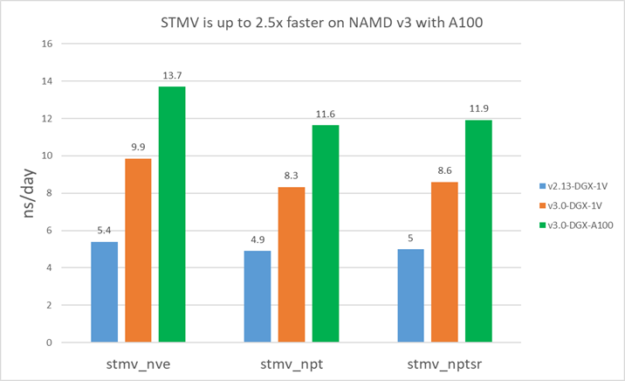
NAMD v3 still runs the atom reassignment between cycles on CPU. There is still a small benefit from using multiple CPU cores. Figure 6 shows the v3 performance using different CPU cores for the Satellite Tobacco Mosaic Virus (STMV) and APOA1 problems.
First, the performance doesn’t change very significantly with different CPU cores. For STMV, using 10 cores shows ~10% benefit compared to one core. However, the benefit plateaus after ~10 cores. This is expected as atom assignment is a small fraction of overall time and there are diminishing returns with more CPU cores.
On the other hand, for the smaller APOA1 problem, using four cores shows ~5% benefit compared to using one core. However, using more than four cores decreases the performance a bit. This is because more CPU threads would lead to more overhead in the GPU computation part. For the smaller APOA1 problem, those overheads can become non-negligible when using more CPU cores.
For large problems like STMV, it’s good enough to use ~10 CPU cores. For small problems like APOA1, it’s best to use only a few CPU cores, such as four.
Performance on a single GPU
For v2.13, because it’s CPU-bound, you use all the available CPU cores. For v3, we used 16 cores for STMV and four cores for APOA1.
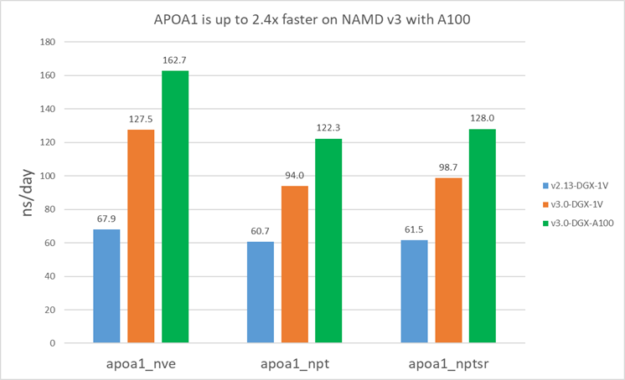
The following results show that v3 is about 1.5-1.9X faster than v2.13 on V100. Furthermore, v3 on A100 is ~1.4X faster than V100 for STMV and ~1.3X faster for APOA1. APOA1 saw smaller benefit on A100 because it cannot fully use all the A100 computing resources due to the much smaller atom count.
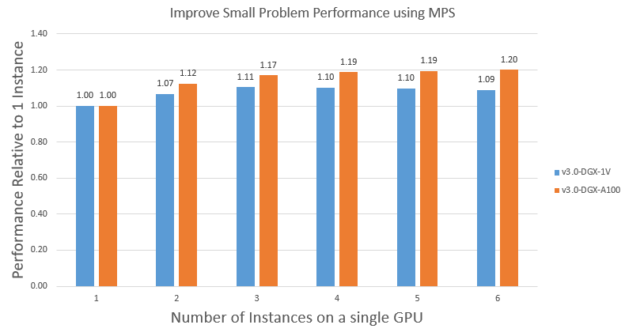
Improve small problem performance using MPS
As we discussed earlier in this post, small problems like APOA1 can’t fully use all the A100 resources. In those cases, you can use MPS to run multiple instances on a single GPU concurrently to improve overall throughput.
To demonstrate this, we use MPS to run multiple instances of the APOA1_NVE problem on a single V100 and A100. Figure 9 shows the scaling on V100 and A100. Running multiple instances using MPS can improve the APOA1_NVE performance by ~1.1X on V100 and ~1.2X on A100. A100 got more benefit because it has more streaming multiprocessors than V100, so it was more under-used. Also because of this, it takes about two instances to saturate the V100 while it takes about three instances to saturate the A100.
It’s also interesting to note that the saturated performance of APOA1_NVE on A100 is ~1.4X of that on V100, which is like the bigger problem STMV, as expected. Finally, it’s worth noting that you could also run a mix of different input problems using MPS to get similar benefits.
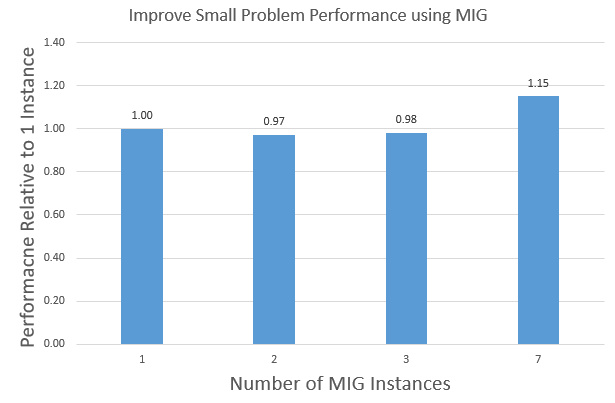
Improve small problem performance using MIG
A100 introduces Multi-Instance GPU (MIG). This provides another way to improve GPU utilization for small problems. MIG partitions the A100 GPU into multiple instances with dedicated hardware resources: compute, memory, cache, and memory bandwidth. This allows for the parallel execution of multiple processes with hardware isolation for predictable latency and throughput.
The main difference between MIG and MPS is that MPS does not partition the hardware resources for application processes. MIG provides more security than MPS, as one process cannot interfere with another process. MIG supports partitioning the A100 GPU into 1/2/3/7 instances. Figure 10 shows the aggregate APOA1_NVE performance of running 1/2/3/7 instances on a single A100 GPU using MIG.
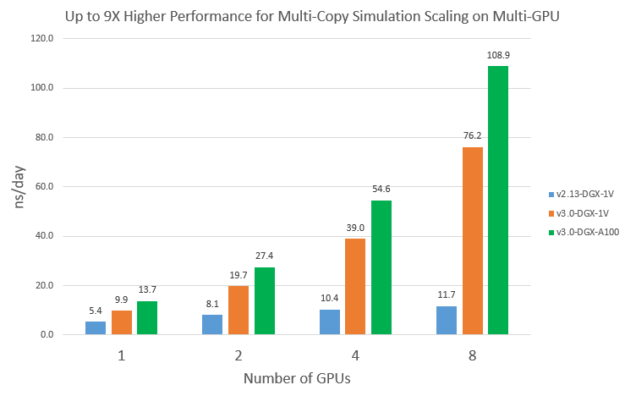
Multi-copy simulation scaling on multi-GPU
Even though the GPU optimizations in v3 only works on single-GPU so far, it does benefit some multi-GPU use cases, such as multi-copy simulations. When you launch multiple replica simulations on a multi-GPU system concurrently, the available CPU cores for each run are reduced compared to running just one simulation. Because NAMD v2.13 is CPU-bound, multi-copy scalability is expected to be not ideal. On the other hand, because v3 performance depends much less on CPU, it would get much better scaling.
To demonstrate this, we launched multiple independent STMV_NVE runs on multiple GPUs concurrently. Each run used all the available CPU cores divided by the number of GPUs. Figure 11 compares the scalability of v2.13 and v3 from one to eight GPUs. You can see that v2.13 doesn’t scale very well: eight V100s is about 2.2x faster than 1 V100. On the other hand, v3 achieved basically a linear speedup on eight V100s and eight A100s. As a result, multi-copy simulation performance of v3 on eight A100s is about 9X higher than v2.13 on eight V100s.
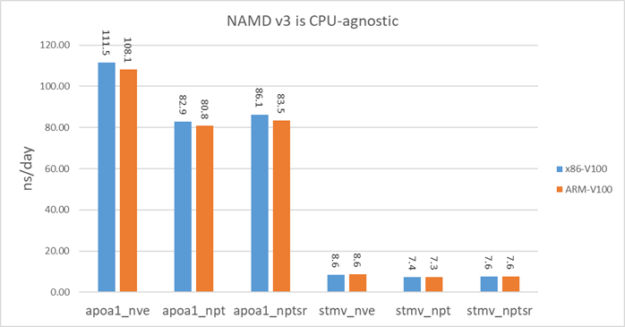
NAMD v3 performance on ARM
NAMD v3 is GPU-bound so it is expected to perform similarly on different CPU architectures. To demonstrate that, we benchmarked v3 on an HPE Apollo 70 system. This system has two ThunderX2 ARM64 28-core CPUs and the GPU is V100-PCIE. V100-PCIE has a lower clock frequency than the V100-SXM2 in DGX-1V systems.
For a fair comparison, we also benchmarked on a x86 system with V100-PCIE. The CPU on this x86 system is Xeon E5-2698 v3. Figure 12 shows that NAMD v3 basically performs equally well on the ARM system as the x86 system.
Next steps
To try out NAMD v3, download the container from NVIDIA NGC.
Benchmark system
We benchmarked NAMD v3 on the NVIDIA DGX-1V and DGX-A100 systems. The DGX-1V system has two Intel Xeon E5-2698 v4 20-core CPUs and eight V100 GPUs. The DGX-A100 system has two AMD Rome 7742 64-core CPUs and eight A100 GPUs.
For more information about changes in NAMD v3, see Scalable molecular dynamics on CPU and GPU architectures with NAMD.
For more information about how the University of Illinois used NVIDIA Nsight Systems to optimize their NAMD Molecular Dynamics Program for GPU acceleration, see GTC 2020: Rebalancing the Load: Profile-Guided Optimization of the NAMD Molecular Dynamics Program for Modern GPUs using Nsight Systems.
