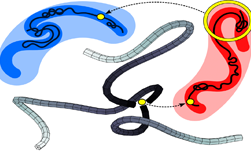
In the last five minutes, you have probably come into contact with more polymers than you can count. In fact, they are everywhere; in grocery bags, water bottles, phones, computers, food packaging, auto parts, tires, airplanes, and toys.
To get a better sense of how polymers behave in different environments Airidas Korolkovas, a postdoctoral researcher at Uppsala University in Sweden, has developed a unique algorithm that can simulate polymers in record time.
Korolkovas’s work focuses on visualizing star polymers, which have been widely used in drug development, material development, tissue engineering, nanoelectronics, and many other applications.
“Star polymers are within the most topologically entangled macromolecules. For a star to move the current theory is that one arm must retract to the branch point. The probability of this event falls exponentially with molecular weight, and a quicker relaxation pathway eventually takes over,” Korolkovas wrote in a post.
“With a simulation over a hundred times faster than earlier studies, it is demonstrated that the mean square displacement scales with a power law 1/16 in time, instead of the previously assumed zero. It suggests that star polymer motion is the result of two linear relaxations coinciding in time,” Korolkovas said.
To achieve the 100x performance improvement, a unique computer algorithm was invented to capture the billions of steps needed for entangled polymers. The algorithm, which was written in CUDA, runs on an NVIDIA TITAN Xp GPU and takes advantage of texture mapping, a feature originally designed for video games.
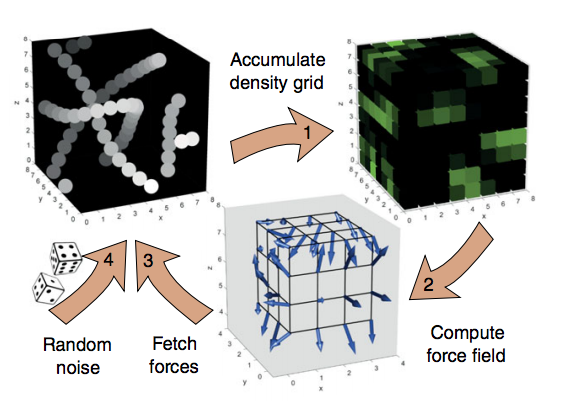
“Here [texture mapping] has been repurposed to calculate the molecular forces inside a little polymer droplet. Using a streamlined physics model, this simulation runs hundreds of times faster than traditional code,” Korolkovas said.
“Using a streamlined physics model, this simulation runs hundreds of times faster than traditional code. It opens new horizons on the time scales that can be addressed in scientific computing. This can further push the frontiers for the latest generation of supercomputers, like the recently opened Summit in Oak Ridge National Lab, USA, which has almost 30,000 GPUs,” Korolkovas said.
A paper describing the method was recently published in the Advanced Theory and Simulations journal. The source code is also publicly available.
Read more>
AI-Generated Summary
- Airidas Korolkovas, a postdoctoral researcher at Uppsala University in Sweden, has developed an algorithm that simulates polymers, specifically star polymers, in record time.
- The algorithm, written in CUDA, runs on an NVIDIA TITAN Xp GPU and achieves a 100x performance improvement by utilizing texture mapping, a feature originally designed for video games, to calculate molecular forces.
- Korolkovas's simulation suggests that star polymer motion results from two linear relaxations coinciding in time, contradicting previous assumptions, and opens new possibilities for scientific computing on the latest generation of supercomputers.
AI-generated content may summarize information incompletely. Verify important information. Learn more