
The upcoming 4.1 release of NVIDIA Parabricks, a suite of accelerated genomic analysis applications, goes further than ever before in accelerating sequencing alignment and increasing the accuracy of deep learning variant calling. The release includes a new workflow for PacBio long-read data, featuring an accelerated Minimap2 tool and Google’s DeepVariant for full GPU-enabled, end-to-end analysis of PacBio data.
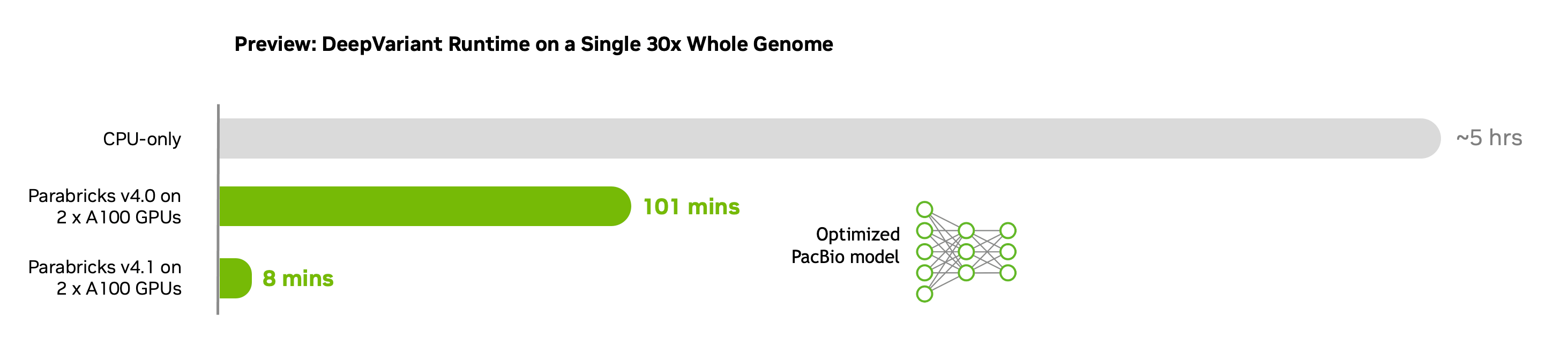
NVIDIA Parabricks is free to use with an option for paid enterprise support. It contains a variety of optimized and AI-based industry-standard genomic tools delivering up to 80x acceleration over CPU-based tools and reducing compute costs by up to 50%. A 30x whole genome can now be analyzed in just 16 minutes compared to ~24 hours on CPU, translating to the analysis of up to 30,000 whole genomes a year on a single server.
A quick look at Parabricks v4.1 features
- A new DeepVariant Re-Training tool, to enable anyone to re-train or fine-tune DeepVariant for their own data, enabling more accurate variant calling (available now on NGC).
- An end-to-end (FastQ-to-VCF) accelerated workflow for PacBio, which will be made available on the Parabricks workflows on GitHub, Terra.Bio, and other cloud platforms.
- New accelerated Minimap2 tool for alignment of PacBio’s long reads.
- New accelerated DeepVariant variant caller for PacBio data with an 8-minute runtime for a 30x whole genome on 2xA100 GPUs.
- Further acceleration of the short-read germline pipeline for a 30x whole genome in 16 mins on a DGX A100 GPU [8xA100 GPUs] compared to 21 mins in v4.0 and ~24 hours on CPU-only.
- Compatibility with the new NVIDIA H100 GPU, which includes a powerful DPX instruction for boosting dynamic programming algorithms like Smith-Waterman for local sequence alignment.
Sign up for notification of the Parabricks 4.1 release, or try the prerelease DeepVariant re-training tool.
Supporting long-read analysis
Long-read sequencing, the capability of sequencing significantly longer fragments of DNA, has multiple inherent advantages over traditional short-read sequencing. Most prominently, the reads are more easily assembled into the full genome.
Lower levels of ambiguity and alignment error make long-read sequencing better for more challenging parts of the genome (for example, highly repetitive regions) or for assembling a genome de novo (without a provided reference).
This has resulted in a multitude of improvements for the sequencing community, including a greater understanding of structural variants (large insertions, deletions, inversions, duplications, and so on). Structural variants can be pathogenic for diseases, such as Lou Gehrig’s disease (ALS), Parkinson’s disease, and cardiac diseases.
It has also finally enabled the scientific community to fully complete the human reference genome end-to-end, known as the telomere-to-telomere (T2T) genome released in 2022.
PacBio is a prominent leader in long-read sequencing. Their technology produces reads up to 25 kilobases in length (compared to short-read sequencing of <300 bases per read). They are also pushing the boundaries of sequencing accuracy with their HiFi read technology based on circular consensus sequencing, and analysis with the transformer-based, deep learning model DeepConsensus.
PacBio’s Revio long-read sequencing system features NVIDIA GPUs and can scale this approach to up to 1,300 human whole genomes per year.
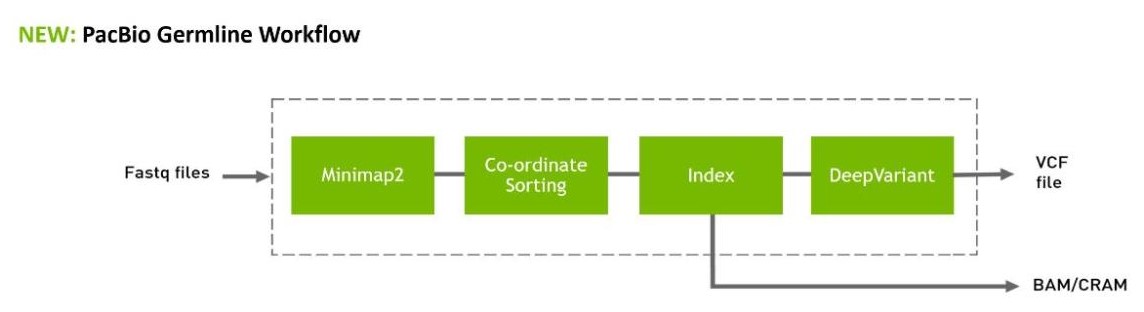
Downstream of this, NVIDIA Parabricks 4.1 software can be used for GPU-accelerated alignment with Minimap2, and variant calling with DeepVariant’s PacBio model, to provide a complete end-to-end workflow on GPU for PacBio data.
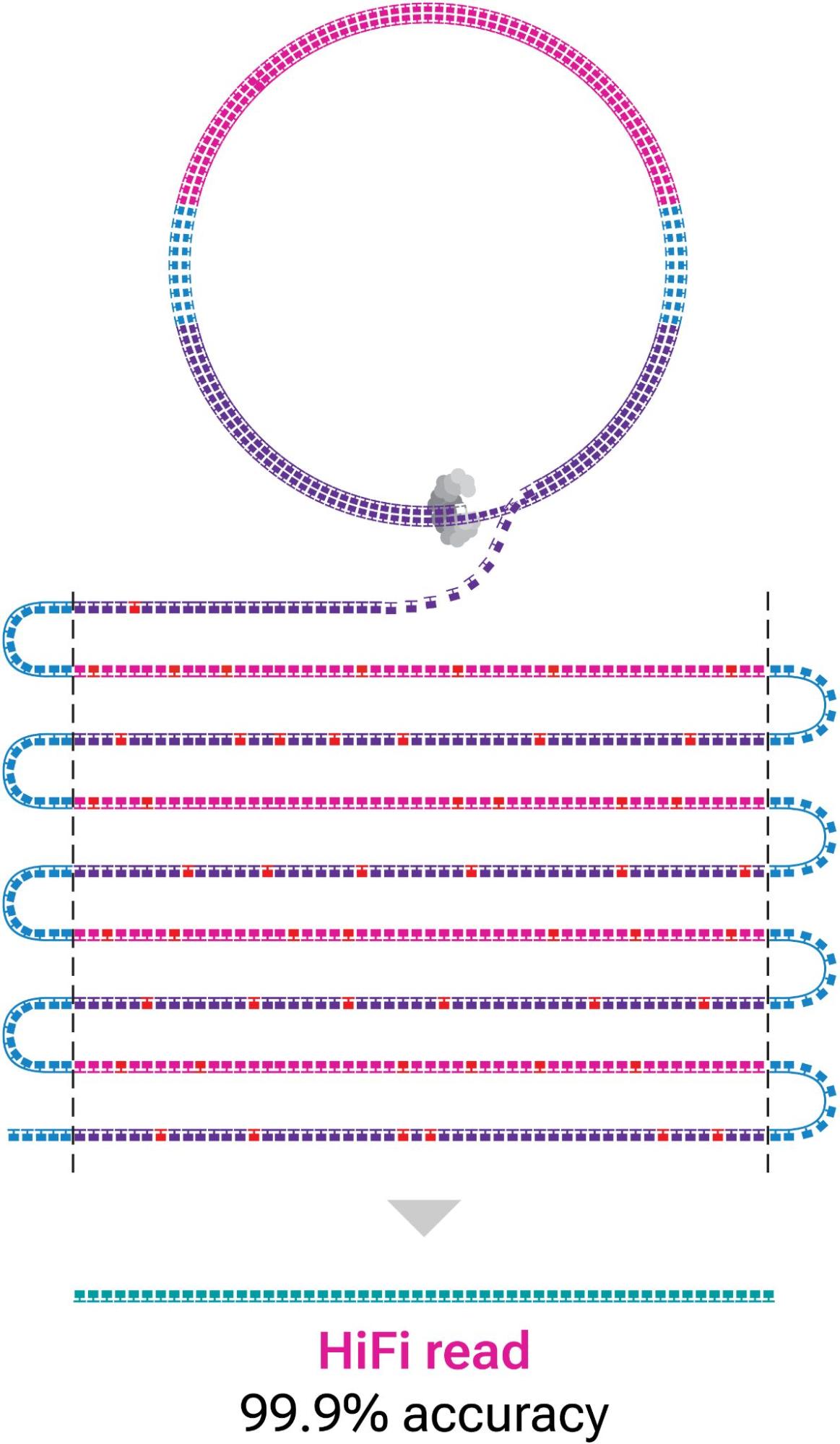
Figure 3 shows that HiFi reads are generated by combining multiple consecutive observations of a DNA molecule (subreads), driving the accuracy of individual HiFi reads over 99%.
DeepVariant re-training with Parabricks
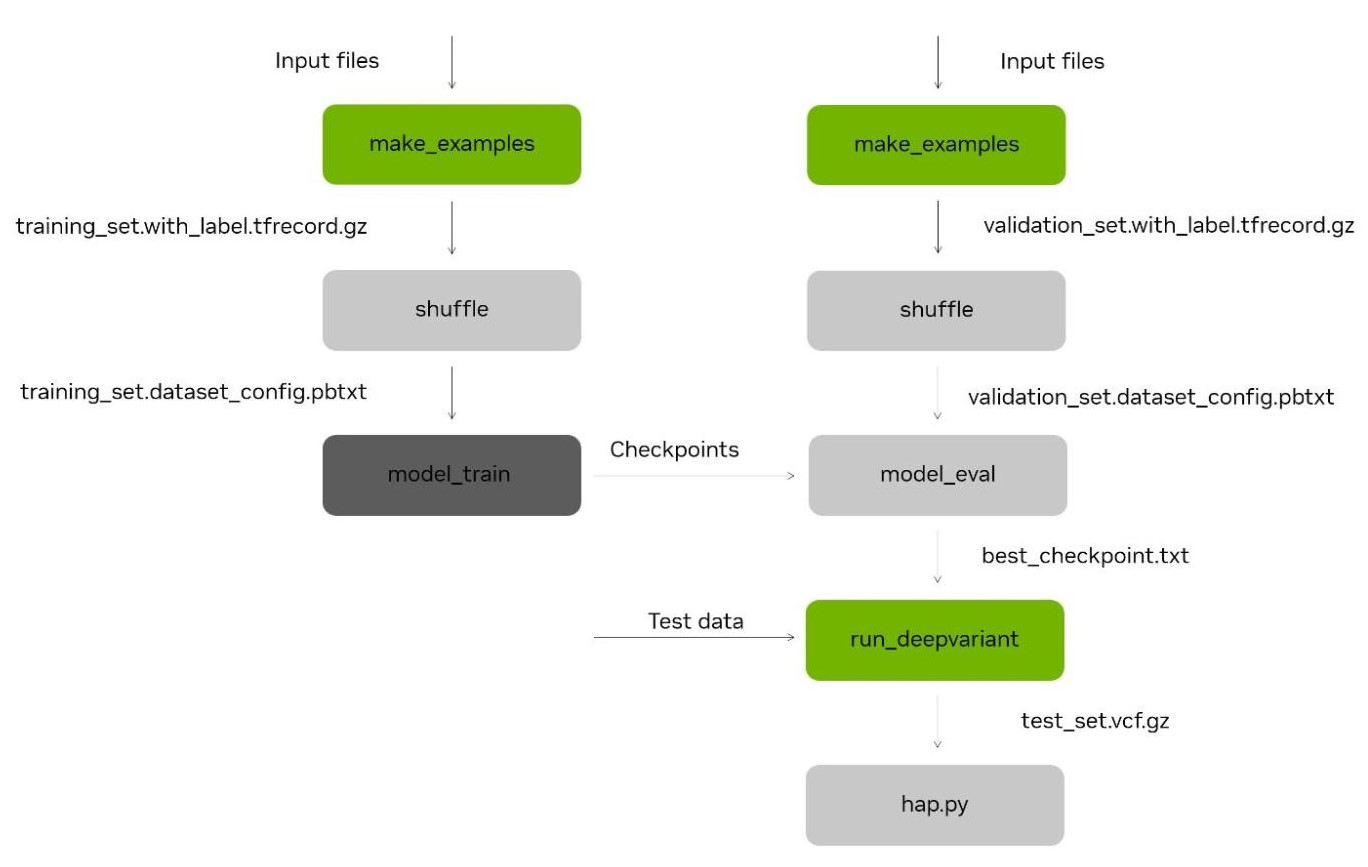
DeepVariant is a CNN-based accurate variant caller for germline workflows of short and long-read data, accelerated on GPUs as part of NVIDIA Parabricks. Parabricks 4.1 includes a framework for re-training and fine-tuning the underlying CNN model to bring more accurate variant calls to analysis workflows.
Specifically, this has the advantage of being able to fine-tune the model to individual datasets and recognize any non-random artifacts produced in the subsequent data. This has been successfully applied at the sequencer level, for example with Ultima, Singular, and PacBio all producing their own specific models trained to their unique error profiles.
It has also been applied at the project level, for example with Regeneron Genetic Center’s exome sequencing as part of the UKBioBank project. Different labs often use different versions of sequencers, wet lab kits, and reagents, and have different lab processes generally. All these differences could introduce subtle and unique artifacts to their samples.
By using the DeepVariant base model as a warm start, implementing lab-specific fine-tuning can be a relatively simple process through sequencing a handful of genome-in-a-bottle cell lines for training, testing, and validating.
In the case of Regeneron, training with a single V100 GPU for 12 hours and on just one sample (HG001) was enough to see the model converge, with 20% of the data held aside for testing and a second sample (HG002) for validation. This led to impressive improvements in accuracy from a relatively small amount of data, for example reducing their Mendelian error rate for indels from 0.075 to 0.056.
Get started with NVIDIA Parabricks
NVIDIA Parabricks is available free on NVIDIA NGC and DGX Cloud, as well as other cloud service platforms and providers. Get started with the new DeepVariant re-training framework today through the Parabricks collection on NGC.
Join us at NVIDIA GTC 2023 and learn more about training DeepVariant models with Parabricks.
For access to other features mentioned in this post, sign up for notification of the Parabricks 4.1 release coming out in Q2. You can learn more about Parabricks on the Accelerated Genome Sequencing Analysis page, including how to purchase enterprise support for Parabricks through NVIDIA AI Enterprise, with guaranteed response times, priority security notifications, and access to AI experts from NVIDIA.
Join our engineers in the Parabricks forum to dive deeper into sequencing and deep variant calling.
