
The human exome is key to understanding and treating genetic disorders and disease. Although the exome consists of just over 1% of the human genome, it also contains approximately 85% of known variants with significant disease-associated mutations. This is why whole-exome sequencing, involving the extraction and sequencing of these regions, is popular in clinical research and practice, where optimizing for accuracy, runtime, and cost is important.
This post demonstrates how NVIDIA Parabricks, a suite of accelerated genomic analysis applications for high-throughput data, can be used for exome analysis. NVIDIA Parabricks significantly reduces both runtime and cost of analysis, while maximizing variant calling accuracy. Whole exome sequencing data analysis can be run on a range of GPUs available both on-premises and in every major cloud provider.
Maximize exome sequencing analysis accuracy with deep learning models
UK Biobank, the world’s most comprehensive publicly available biomedical data resource, provides exome data for 470,000 participants, all sequenced by the Regeneron Genetics Center (RGC). This data is available to researchers worldwide through the UK BioBank Research Analysis Portal, which provides both secure data access and analysis software (such as NVIDIA Parabricks) through DNAnexus.
The human genome contains more than 180,000 protein-coding regions, or exons, which together make up an exome. Each exome contains about 30 million nucleotides. Hence, variant calling is critical in large population studies where even low false positive and negative rates can have sizable impact. To learn more, see Sequencing Your Genome: What Does It Mean?
For this reason, the RGC analyzed UK Biobank exomes with a custom-trained version of Google’s DeepVariant, a high-accuracy deep learning approach to variant classification. This method was accelerated and deployed through NVIDIA Parabricks, providing the exact results as the CPU code, with a faster runtime and lower cost per exome for the RGC.
According to Will Salerno, executive director of RGC Genome Informatics and Data Engineering, “One of the key components of optimizing with Parabricks was not just making it faster and cheaper, but also getting the exact same variants. This reproducibility is critical for us, this point of transparency. We don’t want secret sauce, we want special sauce that works as well for everyone as it does for us. Everything we do, we want to be accessible to any of our partners who could benefit from these methods.”
There are a large variety of variant calling tools available to genomics researchers, from statistical techniques (Bayesian or Gaussian mixture models, for example) to deep learning approaches (convolutional or recurrent neural networks) that classifies exome variants into signal or noise.
Although statistical techniques may provide a more generalizable approach, if the raw data is available to train a deep learning algorithm to a given data type, these models can be incredibly accurate. One example is a genome-in-a-bottle cell line of the same organism/genome, sequenced in the same lab with the same technique and lab protocols.
As such, deep learning variant calling dominated submissions to the most recent precisionFDA Truth Challenge, with 68% of submissions being deep-learning based. Multiple categories won with DeepVariant itself.
DeepVariant uses a convolutional neural network to identify variants in a window of next-generation sequencing (NGS) reads or pile-ups, and includes models for all sequencing platforms, including not only Illumina data, but also PacBio data, Oxford Nanopore data, and the emerging sequencing platforms, whole genome samples, exome samples, and more.
NVIDIA Parabricks offers GPU-accelerated DeepVariant, along with several other variant calling tools. It also includes optimized versions of several such models through TensorRT.
See below for an example of an NVIDIA Parabricks DeepVariant command, as shown in the NVIDIA Parabricks documentation. All NVIDIA Parabricks tools are drop-in replacement commands, enabling the same analysis to run easily on GPU.
# This command assumes all the inputs are in <INPUT_DIR> and all the outputs go to <OUTPUT_DIR>.
$ docker run --rm --gpus all --volume <INPUT_DIR>:/workdir --volume <OUTPUT_DIR>:/outputdir
-w /workdir \
nvcr.io/nvidia/clara/clara-parabricks:4.0.0-1 \
pbrun deepvariant \
--ref /workdir/${REFERENCE_FILE} \
--in-bam /workdir/${INPUT_BAM} \
--out-variants /outputdir/${OUTPUT_VCF}
Using a well-suited model trained on appropriate data can have a significant impact on the subsequent accuracy of variant calls. For example, variant calling exome data using the DeepVariant model trained on whole exome sequencing (WES) data compared to that trained on whole genome sequencing (WGS) data, yields 519 more true positive calls, 42 fewer false positive calls, and 519 fewer false negative calls.
This translates to an increase in F1 score of 1% for single nucleotide polymorphisms (SNPs) and nearly 2% for indels. Results run with NVIDIA Parabricks on genome-in-a-bottle ground truth data are shown in Table 1.
| HG003-WES-100x | Type | Total Loci | True Positives | False Negatives | False Positives | Recall | Precision | F1 Score |
| WES model | INDEL | 1051 | 1020 | 31 | 9 | 0.97050 | 0.99143 | 0.98086 |
| WES model | SNP | 25279 | 24976 | 303 | 46 | 0.98801 | 0.99816 | 0.99306 |
| WGS model | INDEL | 1051 | 1006 | 45 | 31 | 0.95718 | 0.97070 | 0.96389 |
| WGS model | SNP | 25279 | 24471 | 808 | 66 | 0.96804 | 0.99731 | 0.98246 |
The ability to switch DeepVariant to a more suitable model—or even fine-tune the model to specific lab protocols (as Regeneron did for UK Biobank)—is a powerful feature of deep learning-based variant calling.
A new DeepVariant retraining tool is now available in NVIDIA Parabricks v4.1, enabling users to do this quickly and easily on NVIDIA GPUs. You can train the model to recognize any non-random artifacts produced in your data due to different versions of sequencers, wet lab kits, reagents, and so forth.
Cost-effective analysis at increased speeds with equivalent results
Computational analysis of exome data is a stepwise increase in time and cost when compared to small-panel analysis. For clinical exome sequencing analysis, accelerated analysis is important in delivering results at scale.
Agilent Alissa Reporter software, for example, delivers exome analysis leveraging NVIDIA GPUs and NVIDIA Parabricks with autoscaling in the cloud. This means Agilent can deliver clinical insights from genomic data to their customers at lower cost and faster runtime for thousands of samples.
Agilent has reported that their underlying GATK workflows that previously took up to 5 hours and cost as much as $10, have now been reduced to 9 minutes (96% reduction in runtime) and just a few dollars per sample.
“The more information we can derive from a sample, the better,” said Joachim De Schrijver, product owner for Alissa Reporter. “Sequencing whole exomes instead of small panels of genes achieves this well, but the resultant FASTQ files range from five to ten GB per sample, which can mean hours of computation to extract meaningful life-impacting results.”
“Agilent Alissa Reporter leverages GPUs and Parabricks to address this, and processes the data in just minutes. Additionally, this reduces the cost of the underlying infrastructure in the cloud, enabling us to offer highly competitive pricing,” he added.
In addition to accelerating DeepVariant, NVIDIA Parabricks also accelerates GATK best practice versions of variant callers, including HaplotypeCaller (for germline) and Mutect2 (for somatic). In NVIDIA Parabricks, both of these produce equivalent results to the open-source versions (0.999 F1 for both SNPs and Indels respectively), but at faster speeds and lower cost.
As shown in Figure 1, running the NVIDIA Parabricks germline pipelines (BWA-MEM, sort, mark duplicates, BQSR, and HaplotypeCaller or DeepVariant) on a single exome can reduce runtime from over 3 hours (with open-source equivalents on a standard CPU instance), to as little as 11 minutes with DeepVariant (17x faster) and 6.5 minutes with HaplotypeCaller (33x faster) on an instance with eight NVIDIA T4 GPUs.
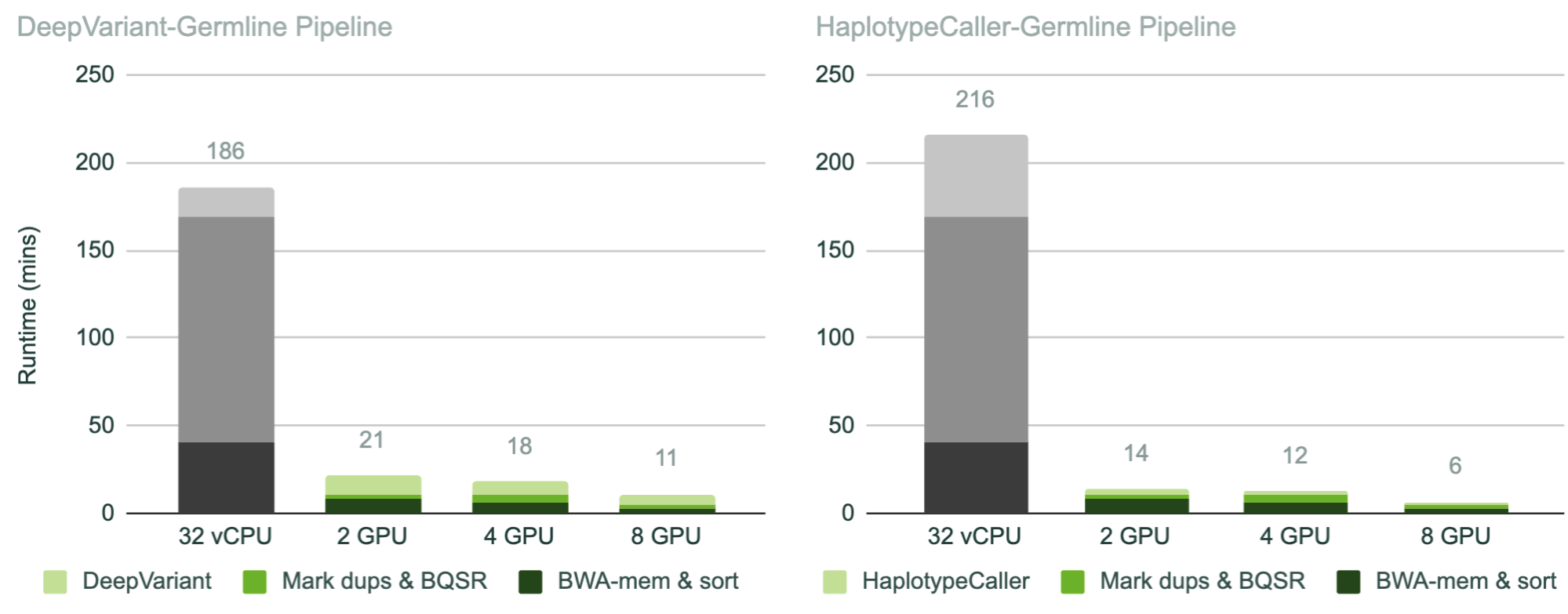
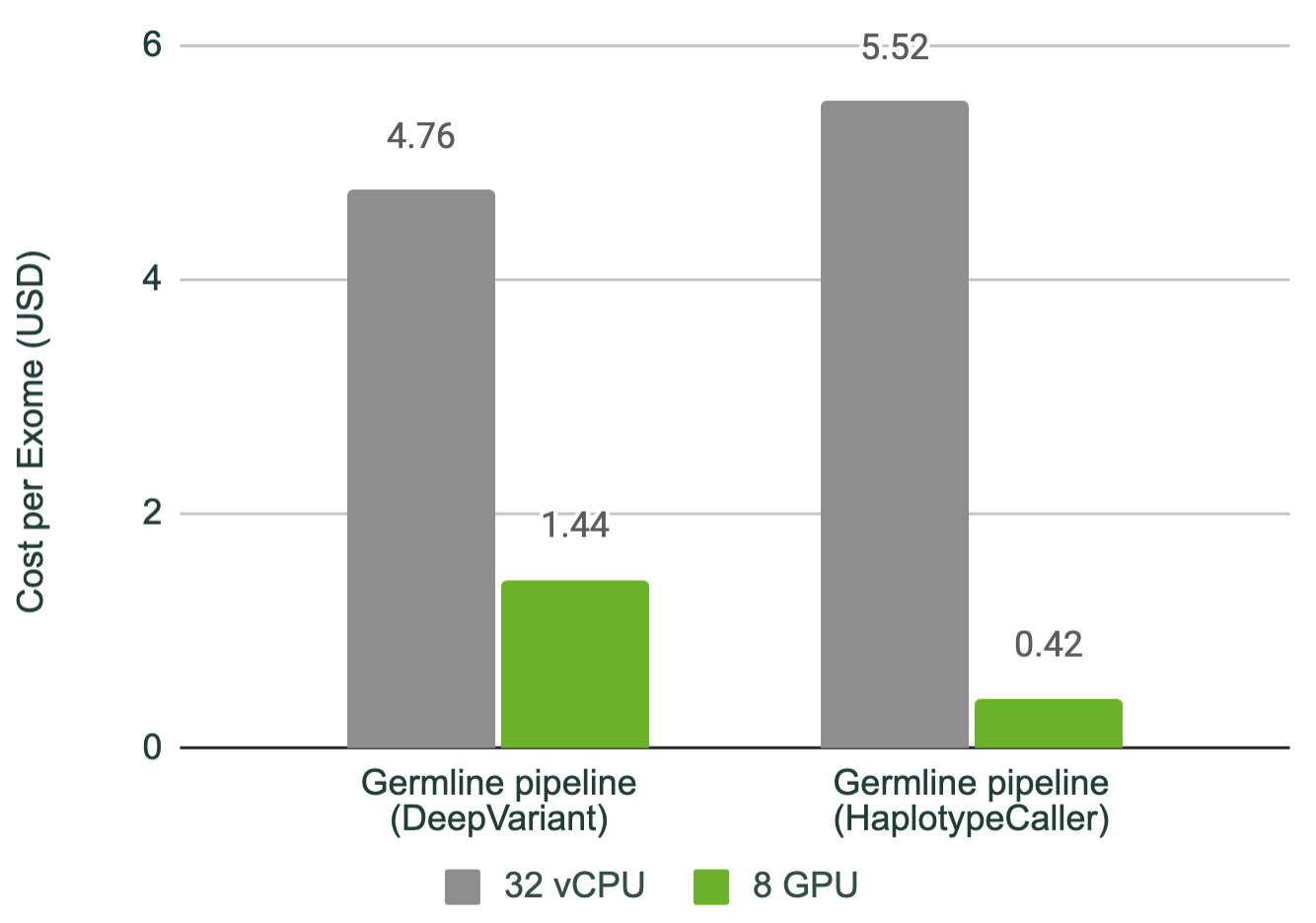
This acceleration factor translates to an impressive cost saving per exome, as the instances are run for shorter periods of time. As shown in Figure 2, running the NVIDIA Parabricks germline pipeline with DeepVariant on eight NVIDIA T4 GPUs reduces the cost from $4.76 to $1.44 (70% less expensive) per sample, and with HaplotypeCaller from $5.52 to just 42 cents (92% less expensive).
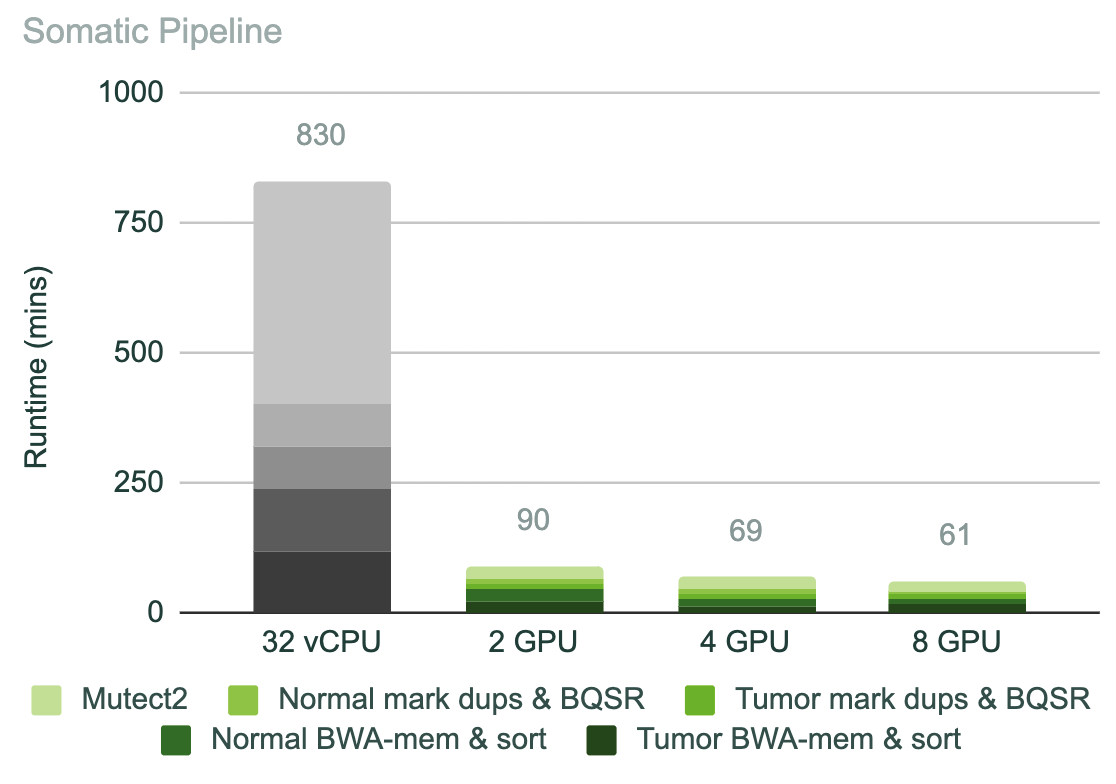
For more complex pipelines, these runtimes can stack to make the analysis step of sequencing a very large bottleneck. For example, in cancer research, where exomes are a common method of sequencing, and where both tumor and normal tissue are often sequenced and to a deeper coverage, a standard exome pipeline for these tumor-normal pairs can take 14 hours to run on a CPU instance. As shown in Figure 3, this can be reduced to 1.5 hours on only two NVIDIA T4 GPUs.
Get started with NVIDIA Parabricks
Download NVIDIA Parabricks for free, including the new DeepVariant retraining tool. For hardware and software requirements and tutorials, see the NVIDIA Parabricks documentation.
Visit the NVIDIA Parabricks page to learn more, including how to purchase enterprise support for access to NVIDIA experts, guaranteed response times, and security notifications.
