单细胞基因组学研究继续推进疾病预防药物的发现。例如,它在为当前的新冠肺炎大流行开发治疗、识别易受感染的细胞以及揭示受感染患者免疫系统的变化方面起着关键作用。然而,随着大规模单细胞数据集可用性的不断提高,计算效率的低下明显影响了科学研究的速度。将这些计算瓶颈转移到 GPU 已经证明了有趣的结果。
在最近的一篇博客文章中, NVIDIA 对 100 万个小鼠脑细胞进行了基准分析,这些脑细胞通过 10 倍基因组学测序。结果表明,在 GCP CPU 实例上运行端到端工作流需要三个多小时,而在单个 NVIDIA V100 GPU 上处理整个数据集只需 11 分钟。此外,在 GCP GPU 实例上运行 RAPIDS 分析的成本也比 CPU 版本低 3 倍。此处阅读博客。
按照Jupyter 笔记本对该数据集进行 RAPIDS 分析。要运行笔记本,文件rapids_scanpy_funcs.py和utils.py必须与笔记本位于同一文件夹中。我们提供了第二个笔记本,其中包含 CPU 版本的分析here。在与 Google Dataproc 团队的合作下,我们构建了一个入门指南,以帮助开发人员快速运行这个转录组学用例。最后,看看这个 NVIDIA 和谷歌云共同撰写博客文章,它展示了工作的影响。
对 GPU 进行单细胞 RNA 分析
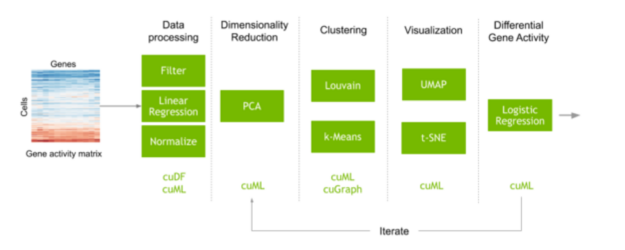
执行单细胞分析的典型工作流程通常从一个矩阵开始,该矩阵映射每个细胞中测量的每个基因脚本的计数。执行预处理步骤以滤除噪声,并对数据进行归一化以获得在数据集的每个单独单元中测量的每个基因的表达。在这一步中,机器学习也常用于纠正数据收集中不需要的伪影。基因的数量通常相当大,这会产生许多不同的变异,并在计算细胞之间的相似性时增加很多噪音。在识别和可视化具有相似基因表达的细胞簇之前,特征选择和降维可以减少这种噪声。这些细胞簇的转录表达也可以进行比较,以了解为什么不同类型的细胞行为和反应不同。
该分析证明了使用RAPIDS加速使用单个 GPU 分析 100 万个细胞的单细胞 RNA 序列数据。然而,实验只处理了前 100 万个细胞,而不是整个 130 万个细胞。因此,在单细胞 RNA 数据的工作流中处理所有 130 万个细胞的时间几乎是单个 V100 GPU 的两倍。另一方面,相同的工作流在单个 NVIDIA A100 40GB GPU 上只需 11 分钟。不幸的是, V100 的性能下降了近 2 倍,主要原因是 GPU 的内存被超额订阅,从而在需要时溢出到主机内存。在下一节中,我们将更详细地介绍这种行为,但需要明确的是, GPU 的内存是扩展的限制因素。因此,更快地处理更大的工作负载需要更强大的 GPU 服务器,如 A100 或/或将处理分散到多个 GPU 服务器上。
将预处理扩展到多个 GPU 的好处
当工作流的内存使用量超过单个 GPU 的容量时,统一虚拟内存( UVM )可用于超额订阅 GPU ,并自动溢出到主内存。这种方法在探索性数据分析过程中是有利的,因为适度的超额订阅率可以消除在 GPU 内存不足时重新运行工作流的需要。
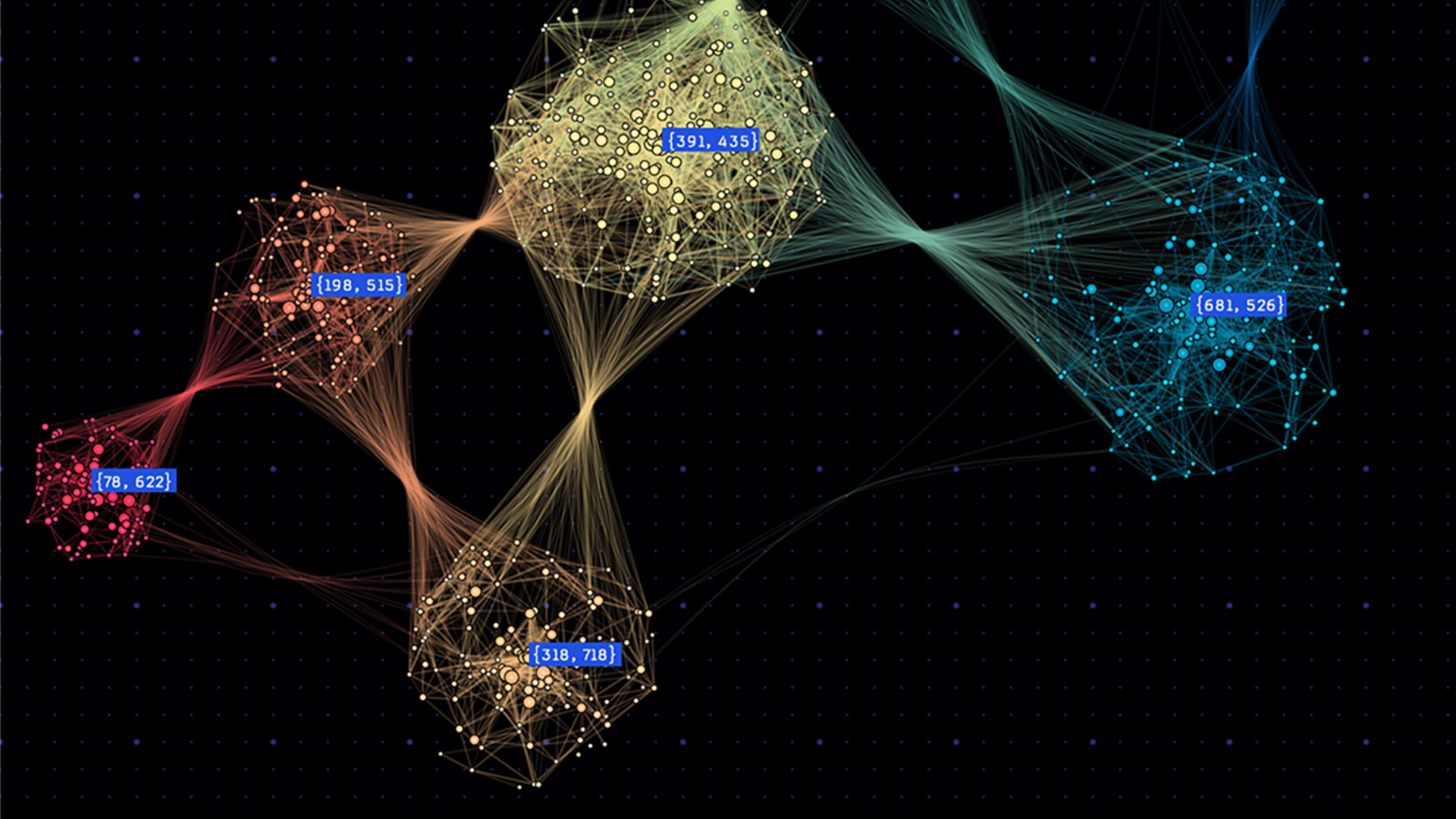
但是,严格依靠 UVM 将 GPU 的内存超额订阅 2 倍或更多可能会导致性能不佳。更糟糕的是,当任何单个计算需要的内存超过 NVIDIA 上的可用内存时,它可能会导致执行无限期挂起。将计算扩展到多个 GPU 可以提高并行性并减少每个 GPU 上的内存占用。在某些情况下,它可以消除超额认购的需要。图 2 表明,我们可以通过将预处理计算扩展到多个 GPU 来实现线性缩放,与单个 GPU V100 GPU 相比, 8 个 GPU s 会产生略微超过 8 倍的加速比。考虑到这一点,需要不到 2 分钟才能将 130 万个细胞和 18k 基因的数据集减少到约 129 万个细胞和 8 GPU上 4k 个高度可变的基因。这超过了 8 . 55 倍的加速,因为单个 V100 需要 16 分钟来运行相同的预处理步骤。
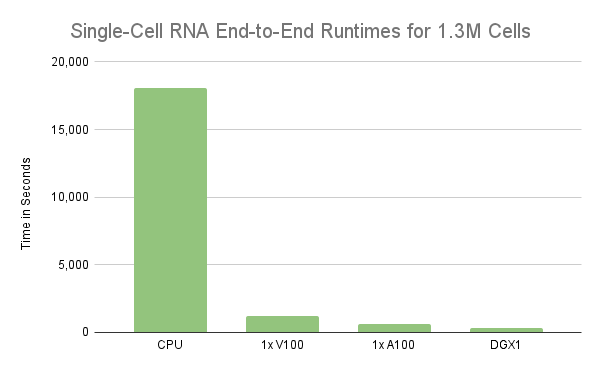
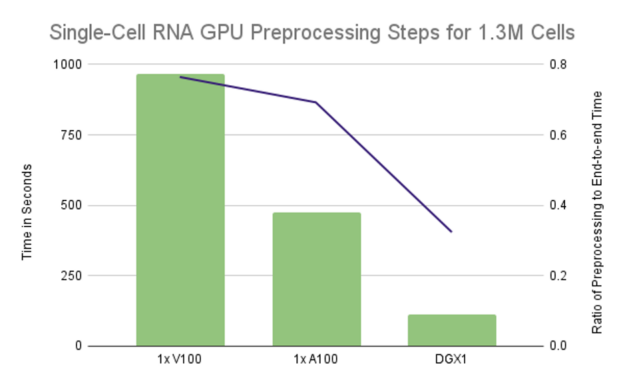
图 3 :单个 GPU 配置的运行时主要由预处理步骤控制,在单个 V100 上占据 75% 的端到端运行时,在单个 A100 上占据 70% 的运行时。利用 DGX1 上的所有 GPU 将比率降低到略高于 32%.
使用 Dask 和 RAPIDS 将单细胞 RNA 笔记本扩展到多个 GPU
许多预处理步骤,如加载数据集、过滤嘈杂的转录本和细胞、将计数标准化为表达式以及特征选择,本质上是并行的,每个 GPU 独立负责其子集。纠正数据收集噪音影响的一个常见步骤是使用不需要的基因(如核糖体基因)的贡献比例,并拟合许多小型线性回归模型,数据集中每个转录本对应一个模型。由于转录本的数量通常可以达到数万个,因此通常使用分散性或可变性的度量方法,只选择几千个最具代表性的基因。
Dask是一个优秀的库,用于在一组工作进程上分发数据处理工作流。 RAPIDS 通过将每个工作进程映射到自己的 GPU ,使 Dask 也能够使用 GPU s 。此外, Dask 提供了一个分布式阵列对象,非常类似于 NumPy 阵列的分布式版本(或CuPy,其 GPU 加速外观相似),它允许用户在多个 GPU 上,甚至跨多台物理机器,分发上述预处理操作的步骤,操作和转换数据的方式与 NumPy 或 CuPy 数组大致相同。
在预处理之后,我们还通过对数据子集进行训练并分配推理来分配主成分分析( PCA )缩减步骤,通过仅将前 50 个主成分恢复到单个 GPU 来降低通信成本,用于剩余的聚类和可视化步骤。该数据集的 PCA 简化单元矩阵仅为 260 MB ,允许在单个 GPU 上执行剩余的聚类和可视化步骤。使用这种设计,即使包含 500 万个单元的数据集也只需要 1GB 内存。
结论
以我们计算工具的发展速度,我们可以假设数据处理量很快就会迎头赶上,特别是对于单细胞分析工作负载,这迫使我们需要更高的扩展。同时,通过将聚类和可视化步骤分布在多个 RAPIDS 上,仍有机会进一步减少探索性数据分析过程的迭代次数。更快的迭代意味着更好的模型,缩短洞察时间,以及更明智的结果。除 T-SNE 外,多 GPU 工作流笔记本的所有集群和可视化步骤都可以通过 GPU cuML 和 cuGraph 分布在 GPU 上的 Dask 工作人员上。