
 Originally trained as a veterinary surgeon, Chris Jewell, a Senior Lecturer in Epidemiology at Lancaster Medical School in the UK became interested in epidemics through his experience working on the foot and mouth disease outbreak in the UK in 2001. His work so far has been on livestock epidemics such as foot and mouth disease, theileriosis, and avian influenza with government organizations in the UK, New Zealand, Australia, and the US. Recently, he has refocused his efforts into the human field where populations and epidemics tend to be larger and therefore need more computing grunt.
Originally trained as a veterinary surgeon, Chris Jewell, a Senior Lecturer in Epidemiology at Lancaster Medical School in the UK became interested in epidemics through his experience working on the foot and mouth disease outbreak in the UK in 2001. His work so far has been on livestock epidemics such as foot and mouth disease, theileriosis, and avian influenza with government organizations in the UK, New Zealand, Australia, and the US. Recently, he has refocused his efforts into the human field where populations and epidemics tend to be larger and therefore need more computing grunt.
Epidemic forecasting centers around Bayesian inference on dynamical models, using Markov Chain Monte Carlo (MCMC) as the model fitting algorithm. As part of this algorithm Chris has had to calculate a statistical likelihood function which itself involves a large sum over pairs of infected and susceptible individuals. He is currently using CUDA technology to accelerate this calculation and enable real-time inference, leading to timely forecasts for informing control decisions.
“Without CUDA technology, the MCMC is simply too slow to be of practical use during a disease outbreak,” he says. “With the 380x speedup over a single core non-vector CPU code, real-time forecasting is now a reality!”
Brad: Can you talk a bit about your current research?
Chris: I work on forecasting for epidemics, using a statistical machine learning framework that aims to track and predict risk from disease outbreaks in real-time. In a disease outbreak response context, I am interested in developing methods to estimate the parameters of stochastic dynamical disease models by fitting them to partially observed epidemic data. Once you know the model parameters, and are satisfied that the model adequately represents the disease process, you can run the epidemic model forward in time to create a forecast—like a weather forecast, but for a disease outbreak.
Bayesian analysis represents the cutting edge for parameter estimation for epidemic models, presenting an elegant way to assimilate previous knowledge of how diseases spread, as well as all sorts of data gathered from the current outbreak. To do this, Markov-Chain Monte Carlo (MCMC) fitting methodology developed over the past 15 years represents the gold standard for numerically estimating Bayesian posterior probability distributions, even for highly non-linear models in extremely high dimensions. For an epidemic in progress, the MCMC is required not only to evaluate the posterior distribution over the model parameters, but must also explore the state space of “cryptic” infections—individuals who have been infected but don’t yet know it. Many hundreds of thousands of iterations are required from the MCMC in order to make an inference, and this must happen quickly for real-time prediction!
MCMC is an inherently serial algorithm, in which the rate-limiting step is the calculation of the Bayesian posterior density function at each iteration. This is akin to an N-body problem, where interactions between pairs of infected-susceptible individuals must be calculated repeatedly as the MCMC explores around the posterior probability distribution. In fact, disease interaction is very much like a gravity model, where transmission of infection is modeled as individuals’ infectivity and susceptibility to disease (c.f. mass) multiplied by a function of distance (network or spatial) between them.
However, unlike a physics research context, for an epidemic forecast to be effective it must be fast! The major challenge is therefore the requirement for massively parallel calculation of the disease interactions, whilst keeping communication between processors to an absolute minimum.
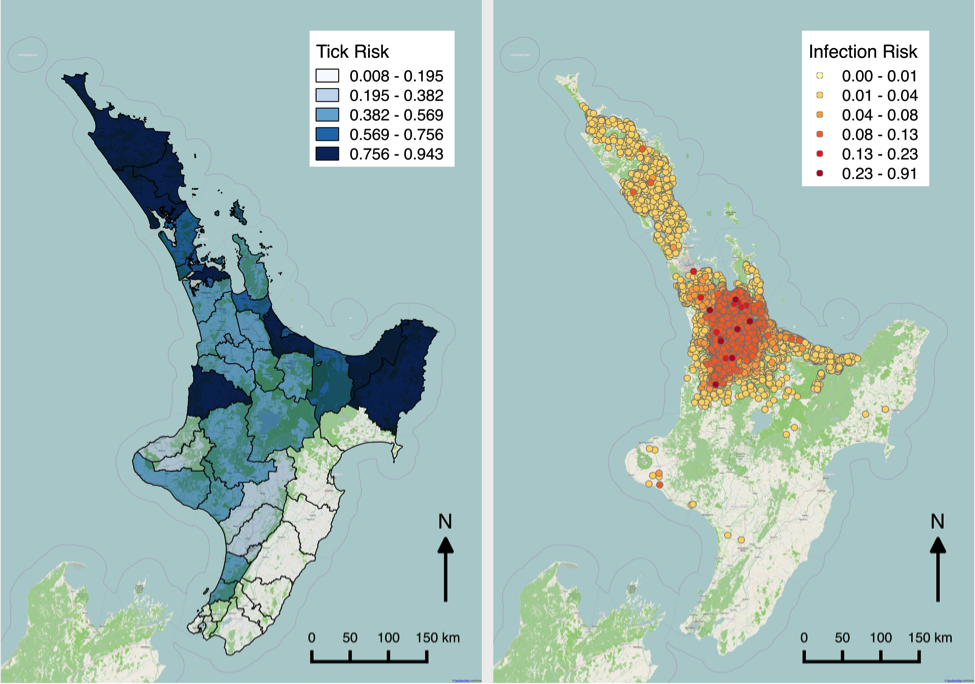
Figure 1 shows maps of my recent work on Theileria orientalis (Ikeda), a tick-borne parasite of cattle, which was introduced into New Zealand in 2012. I have been working with the Ministry for Primary Industries to predict the risk of infection for hither-to uninfected farms, and also the probability of tick occurrence spatially over the country. Given a bit more computing power, this could be humans and malaria!
B: How will focusing on the human field differ from livestock epidemics?
C: There are two main differences between the human and livestock fields. Firstly, we closely manage our livestock: we know where the animals are and when and where they move, and the datasets are consequently highly detailed. For human epidemics, we simply don’t have this quality of data, particularly in developing world situations such as the recent Ebola outbreak, but we are still required to provide the same level of detail in our forecasts. Secondly, livestock populations are generally relatively small, from a few thousand to a few hundred thousand individuals. For humans, increase this by many orders of magnitude, especially when considering pandemic diseases such as influenza!
B: What are some of the biggest challenges you are tackling?
C: Without a doubt, the biggest challenge we face is how to scale forecasting models to ever larger populations, and ever more complex models, which can be tailored to specific disease applications in the blink of an eye. This is where epidemics differ from weather forecasting: disease processes simply do not obey mass action principles, as shown by Bartlett in 1975. Whilst fast, deterministic models of disease are useful for studying basic mathematical principles of epidemics, their use in large pandemic situations is limited due to local stochastic infection events having far-reaching consequences for the wider population.
From a computing perspective, we currently face two problems in epidemic forecasting. The first is how to handle massive amounts of data, whilst maintaining computational feasibility. The second is that as populations get larger, our capacity to collect reliable and repeatable data decreases. Crowd-sourced data is a really exciting prospect for human disease data, but as recent events with Google Flu Trends have shown, simply collecting more data without considering its biases may lead to incorrect predictions. Statistical methods will become ever more complex to account for and correct these biases and uncertainties, but will inevitably have a voracious demand for massively parallel computing with close to negligible communication time.
A second challenge we face is how to make these new developments in forecasting methodology flexible enough to be adapted quickly to new situations. As any scientific programmer knows, an algorithm tailored to a specific problem on specific hardware will outperform a general one-size-fits-all algorithm. However, in our situation, these specializations and/or generalizations occur in the pairwise disease interaction model—the most time-critical steps of our calculations. We are thinking hard about the best way to do this in a software package aimed at epidemiological researchers, rather than specialist statisticians and computer scientists.
B: How are you using GPUs?
C: GPU computing, particularly using CUDA, has revolutionized my research. Whereas previously my algorithms took days to run, I can now run analyses on an overnight time-frame – collecting data at the end of one day, and providing a disease forecast at the beginning of the next.
I’ve explored the whole range of parallel computing in my research, seeking a solution to parallel computation of infected-susceptible disease interactions. Firstly, distributed computing with MPI foundered due to the effect of Amdahl’s Law relating to communication overhead, and difficulties in scheduling massively parallel jobs on busy university HPC systems. Secondly, shared memory implementations had low-communication overhead, but were too limited in terms of compute cores given our research budget.
For me, GPU technology represents the best of both worlds: massively parallel computation with low communication. Plus, it fits under my desk!
B: What NVIDIA technologies are you using?
C: I have mostly been working on systems with two or more NVIDIA Tesla C2075 cards, in both workstation and cluster settings. Despite being relatively old technology, these cards have proved really reliable and effective as accelerating my research!
The CUDA runtime API currently forms the bedrock of my C++ codebase, as it provides an excellent balance between being library rich, and offering the opportunity to hand-optimize algorithms if needed.
B: Which CUDA features and GPU-accelerated libraries do you use?
C: I currently use an intricate web of hand-coded CUDA kernels, calls to cuBLAS, cuSPARSE, cuRAND, and CUDPP. I’ll talk about each separately:
- CUDA / cuSPARSE: I make extensive use of hand-coded kernels for computation. I realized that a huge speed saving could be obtained by using sparse matrix techniques where each non-zero element is a disease interaction term. For this calculation, cuSPARSE didn’t quite do what I needed. However, the documentation on how it works allowed me to use the basic principles on which cuSPARSE is founded to optimize my own approach. I also make use of page-locked memory for accelerating the return of calculation results to the host.
- cuRAND: This goes without saying for a stochastic algorithm implemented on a GPU! The MCMC progresses by making decisions based on random number generation. cuRAND has enabled me to port more of my CPU code to the GPU, allowing me to cut down on memory transfers even for serial portions of code.
- CUDPP: Not an NVIDIA library of course, but an excellent project with an important and burgeoning following! I originally implemented standard routines, such as parallel reduction, in Thrust. During development, this was fast and moderately effective, and allowed me to concentrate on getting my hand-coded kernels correct. However intermediate data allocation in Thrust became a bottleneck. CUDPP, on the other hand, provides a simple (if a little clunky) interface to delegate algorithm configuration to initialization code, cutting down on redundant memory allocations in performance critical areas of the algorithm.
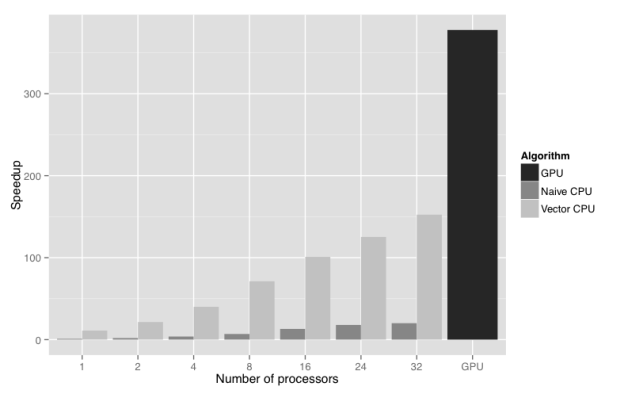
Figure 2 shows the results of a scalability study performed using the UK 2001 foot and mouth outbreak dataset and model (Keeling et al. 2001). The figure compares the wall clock time for GPU, non-vectorised CPU, and vectorised CPU implementations of the Bayesian posterior density function calculation. The test system was an HP Z820 workstation with two Intel Xeon E5-2650 processors, and two NVIDIA Tesla C2075 GPUs, though only one GPU was used. The GPU offers a 380 times speedup of the calculation compared to non-vectorised CPU implementation running with a single CPU core. Moreover, it outperforms by a factor of 2.5 the vectorised CPU version running with all 32 logical CPU cores present on an HP Z820 workstation. The result is that overnight forecasting of another outbreak of foot and mouth disease in the UK is now possible, and can quickly be put into action if the need arises.
B: What types of parallel algorithms have you implemented?
C: My approach to fitting epidemic models to data makes use of Markov Chain Monte Carlo techniques to simulate from complex, high dimensional probability distributions in Bayesian statistics. MCMC itself is an iterative algorithm, and is therefore inherently serial. At each iteration, the MCMC selects a parameter to update, proposes a change in the value of the parameter, and then either accepts or rejects this value based on the value of the posterior distribution function. It is the calculation of the posterior that presents an opportunity for parallelization – a parallel-within-serial algorithm. However, this algorithm is extremely sensitive to Amdahl’s Law, making the GPU an obvious choice as a co-processor.
B: What do you think is most interesting for developers to learn from your project?
C: GPUs can be used as very effective co-processors, even in serial iterative algorithms. A bit of careful thought about code efficiency can pay dividends. Highly efficient and portable implementations of complex algorithms can be made to run orders of magnitude faster than hurriedly coded, problem-focused research code, and saves time in the long run.
B: Any tips for beginning GPU developers?
C: Sure, my top three are:
- Read the CUDA documentation carefully, and accept that learning CUDA will take time. The time’s a good investment though, and pays ample dividends further down the line.
- Make good use of C++ template classes to avoid code bloat. A trap for new CUDA developers is that if you’re not careful, you double the number of lines of code in your software, obscuring readability and increasing the opportunity for bugs to appear.
- Error check. Error check. Error check! Oh, and error check! Be defensive – check the GPU error status after each kernel launch or memory operation, use lots of assert()s, use macros to remove debug code when you’re happy your algorithm implementation is correct.
B: What are you most excited about, in terms of advancements over the next decade?
C: I’m looking forward to the unification of massively parallel numerically oriented processing with standard CPU techniques. A huge amount of progress has been made to simplify the task of managing data between host and device, and easing the code bloat that CUDA has traditionally involved. I look forward to a convergence of parallel and serial computing both in the machine hardware, and also in the software APIs!