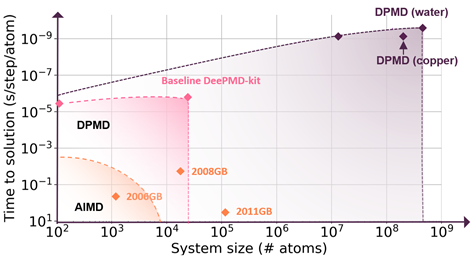
Machine learning interatomic potentials (MLIPs) are transforming the landscape of computational chemistry and materials science. MLIPs enable atomistic simulations that combine the fidelity of computationally expensive quantum chemistry with the scaling power of AI.
Yet, developers working at this intersection face a persistent challenge: a lack of robust, Pythonic toolbox for GPU-accelerated atomistic simulation. For use cases such as running a large number of simultaneous, GPU-accelerated simulations, robust and well-supported tools are either missing in the current software ecosystem or are fragmented across several open source software tools.
Over the past few years, available software for running atomistic simulations with MLIPs has been CPU-centric. Core operations such as neighbor identification, dispersion corrections, long-range interactions, and their associated gradient calculation have traditionally supported only CPU computation, which often struggles to deliver the speed that contemporary research demands. High-throughput simulations of small- to medium-sized atomic systems quickly become bottlenecked by inefficient GPU usage in hybrid workflows where the model is GPU-accelerated in PyTorch but the simulation tooling is serial and CPU-based.
While developers have attempted to implement these operations directly in PyTorch over the years, the general-purpose design of PyTorch leaves performance on the table for the specialized spatial and force calculation operations required in atomistic simulation. This fundamental mismatch between PyTorch capabilities and the demands of atomistic modeling raises an important question: What’s needed to bridge this gap?
NVIDIA ALCHEMI (AI Lab for Chemistry and Materials Innovation), announced at Supercomputing 2024, provides chemistry and materials science developers and researchers with domain-specialized toolkits and NVIDIA NIM microservices optimized on NVIDIA accelerated computing platforms. It is a collection of high-performance, batched and GPU-accelerated tools specifically for enabling atomistic simulations in chemistry and materials science research at the machine learning framework level.
NVIDIA ALCHEMI delivers capabilities across three integrated layers:
- ALCHEMI Toolkit-Ops: A repository of GPU-accelerated, batched common operations for AI-enabled atomistic simulation tasks, such as neighbor list construction, DFT-D3 dispersion corrections, and long-range electrostatics.
- ALCHEMI Toolkit: A collection of GPU-accelerated simulation building blocks, including geometry optimizers, integrators, and data structures to enable large-scale, batched simulations leveraging AI.
- ALCHEMI NIM microservices: A scalable layer of cloud‑ready, domain‑specific microservices for chemistry and materials science, enabling deployment and orchestration on NVIDIA‑accelerated platforms.
This post introduces NVIDIA ALCHEMI Toolkit-Ops, the accelerated batched common operations layer of ALCHEMI. ALCHEMI Toolkit-Ops uses NVIDIA Warp to accelerate and batch common operations in AI-driven atomistic modeling. These operations are exposed through a modular PyTorch accessible API (with a JAX API targeted for a future release) that enables rapid iteration and integration with existing and future atomistic simulation packages.
Figure 1 shows the accelerated batched common operations for atomistic simulations included in this initial release of ALCHEMI Toolkit-Ops. This beta release includes two versions of neighbor lists (naive and cell), DFT-D3 dispersion correction, and long-range coulombic (Ewald and Particle Mesh Ewald) functions.

Figure 2 demonstrates the performance of accelerated kernels in ALCHEMI Toolkit-Ops versus popular kernel-accelerated models like MACE (cuEquivariance) and TensorNet (Warp) to achieve fully parallelized performance and scalability. The blue MLIP baseline allows comparison with advanced features like neighbor lists, dispersion corrections (DFT-D3) and explicit electrostatics computations (Ewald and Particle-Mesh Ewald (PME)). Test systems consisted of ammonia clusters of increasing size packed into various cells using Packmol. Timing results were averaged over 20 runs on an NVIDIA H100 80 GB GPU. The DFT-D3 calculation does not include 6Å due to the long-range nature of D3.
![Benchmarks showing the speed of ALCHEMI Toolkit-Ops neighbors list (both naive O(N²) and cell list O(N) implementations), and DFT-D3 correction and two versions of Ewald summation for electrostatic interactions. All methods are compared to the computational cost of popular kernel-accelerated MLIPs. Left-side panels outline_ batch scaling for fixed number of atoms and variable system size x [batch size], while right-side panels demonstrate timings for a single system growing in size.](https://developer-blogs.nvidia.com/wp-content/uploads/2025/12/deep-dive-Fig2-update-png.webp)
ALCHEMI Toolkit-Ops ecosystem integration
ALCHEMI Toolkit-Ops is designed to integrate seamlessly with the broader PyTorch-based atomistic simulation ecosystem. We are excited to announce in-progress integrations with leading open source tools in the chemistry and materials science community: TorchSim, MatGL, and AIMNet Central.
TorchSim
TorchSim, a next-generation open source atomistic simulation engine, is adopting ALCHEMI Toolkit-Ops kernels to power its GPU-accelerated workflows.TorchSim is a PyTorch-native simulation engine purpose-built for the MLIP era, enabling batched molecular dynamics and structural relaxation across thousands of systems simultaneously on a single GPU. TorchSim will leverage our optimized neighbor lists to drive high-throughput batched operations without sacrificing flexibility or performance.
MatGL
MatGL (Materials Graph Library) is an open source framework for building graph-based machine learning interatomic potentials and foundation potentials for inorganic, molecular, and hybrid materials systems. By integrating ALCHEMI Toolkit-Ops, MatGL significantly accelerates graph-based treatments of long-range interactions, enabling large-scale atomistic simulations that are both faster and more computationally efficient without compromising accuracy.
AIMNet Central
AIMNet Central is a repository for AIMNet2, a general-purpose MLIP capable of modeling neutral, charged, organic, and elemental-organic systems with high fidelity. AIMNet Central is leveraging ALCHEMI Toolkit-Ops to further enhance the performance of its flexible long-range interaction models. Using NVIDIA-accelerated DFT-D3 and neighbor list kernels, AIMNet2 can deliver even faster atomistic simulations for large and periodic systems without compromising accuracy.
How to get started with ALCHEMI Toolkit-Ops
Getting started with ALCHEMI Toolkit-Ops is simple and designed with ease of use in mind.
System and package requirements
- Python 3.11+
- Operating System: Linux (primary), Windows (WSL2), macOS
- NVIDIA GPU (A100 or newer recommended), CUDA compute capability ≥ 8.0
- CUDA Toolkit 12+, NVIDIA driver 570.xx.xx+
Installation
To install ALCHEMI Toolkit-Ops, use the following snippet:
# Install via pip wheel
pip install nvalchemi-toolkit-ops
# Make sure it is importable
python -c "import nvalchemiops; print(nvalchemiops.__version__)"
See the ALCHEMI Toolkit-Ops documentation for other installation instructions. Explore the examples directory in the GitHub repository and run them to test acceleration on your own hardware.
Typical troubleshooting tips:
- Verify CUDA installation and device availability:
nvidia-smi,nvcc --version - Ensure compatible Python version:
python --version - Upgrade dependencies as needed:
pip list | grep torchandpip list | grep warp
Feature highlights
This section dives into three ALCHEMI Toolkit-Ops initial features: high-performance neighbor lists, DFT-D3 dispersion corrections, and long-range electrostatic interactions.
Neighbor lists
Neighbor list construction is the backbone of atomistic simulations enabling calculation of energies and forces with local or semi-local MLIPs. ALCHEMI Toolkit-Ops delivers state-of-the-art GPU performance in PyTorch, achieving performance scaling to millions of atoms per second for batches of many small to medium atomic systems or single large atomic systems.
Capabilities
- Both O(N) (cell list) and O(N²) (naive) algorithms with batched processing
- Periodic boundary support for triclinic cells with arbitrary cell dimensions and partial periodicity
- Supports end-to-end compute graph compilation
- Direct API compatibility with PyTorch
API example
import torch
from nvalchemiops.neighborlist import neighbor_list
# Water molecule
water_positions = torch.tensor([
[0.0, 0.0, 0.0], # O
[0.96, 0.0, 0.0], # H
[-0.24, 0.93, 0.0], # H
], device="cuda", dtype=torch.float32)
# Ammonia molecule (NH3)
ammonia_positions = torch.tensor([
[0.0, 0.0, 0.0], # N
[1.01, 0.0, 0.0], # H
[-0.34, 0.95, 0.0], # H
[-0.34, -0.48, 0.82], # H
], device="cuda", dtype=torch.float32)
# Concatenate positions for batch processing
positions = torch.cat([water_positions, ammonia_positions], dim=0)
# Create batch indices (0 for water, 1 for ammonia)
batch_idx = torch.cat([
torch.zeros(3, dtype=torch.int32, device="cuda"), # Water
torch.ones(4, dtype=torch.int32, device="cuda"), # Ammonia
])
# Define cells for each molecule (large enough to contain them without PBC)
cells = torch.stack([
torch.eye(3, device="cuda") * 10.0, # Water cell
torch.eye(3, device="cuda") * 10.0, # Ammonia cell
])
# non-periodic molecule case
pbc = torch.tensor([
[False, False, False], # Water
[False, False, False], # Ammonia
], device="cuda")
# Cutoff distance in Angstroms
cutoff = 4.0
# Compute neighbor list; here we explicitly request a batched cell list algorithm
neighbor_matrix, num_neighbors, shift_matrix = neighbor_list(
positions, cutoff, cell=cells, pbc=pbc, batch_idx=batch_idx, method="batch_cell_list"
)
print(f"Neighbor matrix: {neighbor_matrix.cpu()}") # [7, num_neighbors.max()]
print(f"Neighbors per atom: {num_neighbors.cpu()}") # [7,]
print(f"Periodic shifts: {shift_matrix.cpu()}")
DFT-D3 dispersion corrections
Realistic molecular modeling must fully account for van der Waals interactions, which standard DFT functionals do not account for systematically. DFT-D3 uses empirical pairwise corrections, leading to substantial improvements in binding energies, lattice structures, conformational analysis, and adsorption studies for common DFT functionals.
Capabilities
- Becke-Johnson (BJ) rational damping variant
- Supports batched and periodic calculations
- Supports smoothing at cutoff distance
- Joint energy, forces, and virial calculation
API example
from nvalchemiops.interactions.dispersion import dftd3
batch_ptr = torch.tensor([0, 3, 7], dtype=torch.int32, device="cuda")
atomic_numbers = torch.tensor(
[6, 1, 1, 7, 1, 1, 1], dtype=torch.int32, device="cuda"
)
# For this snippet, assume d3_params is loaded as:
# d3_params = D3Parameters(rcov=..., r4r2=..., c6ab=..., cn_ref=...)
# Users can refer to the documentation to source DFT-D3 parameters
# and understand the expected data structure
d3_params = ...
# call the DFT-D3 functional interface
energy, forces, coordination_numbers = dftd3(
positions=positions,
numbers=atomic_numbers,
a1=0.3981, a2=4.4211, s8=0.7875, # PBE parameters
neighbor_matrix=neighbor_matrix,
neighbor_matrix_shifts=shift_matrix,
batch_idx=batch_idx,
d3_params=d3_params
)
print(f"Energies: {energy.cpu()}") # [2,]
print(f"Forces: {forces.cpu()}") # [7, 3]
Limitations
The current implementation computes two-body terms only (C6 and C8). Three-body Axilrod-Teller-Muto (ATM/C9) contributions are not included. This generally leads to some over-estimation of dispersion energies.
Long-range electrostatic interactions
Accurate modeling of electrostatic interactions is critical for simulations involving ions/charged species and polar systems. Currently, the most common approach for MLIPs is to learn Coulomb interactions within the short-ranged model. Systematic underestimation of long-range Coulombic effects leads to loss of accuracy in binding energies, solvation structures, and interfacial phenomena.
ALCHEMI Toolkit-Ops provides fully GPU-accelerated Ewald summation methods—both standard Ewald and particle mesh Ewald—enabling GPU-accelerated, efficient and accurate treatment of long-range electrostatics in PyTorch.
For large periodic systems, Ewald-based methods separate electrostatic interactions into short-range and long-range components, each computed in the domain best suited for performance. ALCHEMI Toolkit-Ops provides a dual-cutoff strategy that dramatically reduces redundant neighbor queries and memory overhead compared to naive all-pairs approaches, making high-throughput simulations of charged systems practical on modern GPUs. Users can choose between standard Ewald for smaller systems or PME for larger periodic systems, depending on their specific performance and accuracy needs.
Capabilities
- Ewald summation method
- Particle Mesh Ewald (PME) using B-splines
- Supports batched and periodic systems
- GPU-optimized computation, leveraging cuFFT for fast reciprocal-space evaluation
- PyTorch integration provides native tensor support for end-to-end differentiable workflows
API example
from nvalchemiops.interactions.electrostatics import particle_mesh_ewald
# charges for each atom are randomly generated here
atomic_charges = torch.randn(
positions.size(0), dtype=torch.float32, device="cuda"
)
# compute energy and forces with particle mesh ewald
energy, forces = particle_mesh_ewald(
positions,
atomic_charges,
cells,
alpha=0.3, # adjust Ewald splitting parameter
batch_idx=batch_idx,
neighbor_matrix=neighbor_matrix,
neighbor_matrix_shifts=shift_matrix,
compute_forces=True
)
print(f"Energy: {energy.cpu()}") # [2]
print(f"Forces: {forces.cpu()}") # [7, 3]
Dive deeper into ALCHEMI Toolkit-Ops
ALCHEMI Toolkit-Ops empowers the community with high-performance, accessible atomistic modeling tools on NVIDIA GPUs. To accelerate your chemistry and materials science simulations, visit the NVIDIA/nvalchemi-toolkit-ops GitHub repo and NVIDIA ALCHEMI Toolkit-Ops documentation. You can also explore the examples gallery. This beta release of ALCHEMI Toolkit-Ops focuses on highly efficient neighbor lists, dispersion corrections, and long-range electrostatics. Stay tuned for new features and performance optimizations in future releases.
Acknowledgments
We’d like to thank Professor Shyue Ping Ong; Professor Olexandr Isayev; and the TorchSim committee members Abhijeet Gangan, Orion Archer Cohen, Will Engler, and Ben Blaiszik for working with us to adopt NVIDIA ALCHEMI Toolkit-Ops into their open source projects. We also thank Wen Jie Ong, Piero Altoe, and Kibibi Moseley from NVIDIA for their help preparing this blog post.