几十年来,生物学中最深奥的谜题之一,便是氨基酸链如何自发折叠成复杂而精密的生命结构。尽管研究人员精心构建了各种模拟与统计模型,致力于揭示其中的规律,却始终未能实现对折叠结果的大规模准确预测。
随后,深度学习彻底改变了这一局面。通过直接从序列数据中学习进化的语言规律,人工智能开始揭示分子结构中隐藏的规则,使结构预测从一门艺术转变为一门工程学科。
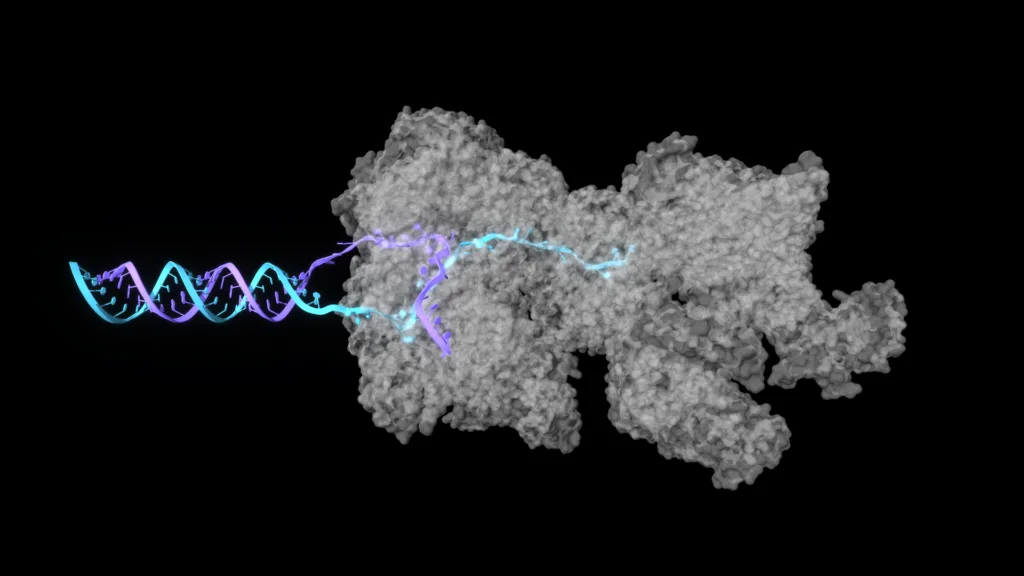
如今,这一转变迎来了一个新的里程碑。由 OpenFold 联盟 开发、NVIDIA 加速支持的 OpenFold3,将生产级蛋白质人工智能引入 NVIDIA 生态系统,实现了开放科学与企业级性能的深度融合。OpenFold3 将结构预测的能力从单一蛋白质拓展至多链复合物、核酸以及小分子配体,全面覆盖生物相互作用的各类关键组分。
借助 NVIDIA cuEquivariance 实现对称感知的 GPU 加速,结合支持 MMseqs2-GPU 实现快速序列搜索,并利用 NVIDIA FLARE 支持联合训练,OpenFold3 可为全球生物制药与生物技术团队提供卓越的速度、可扩展性以及具备隐私保护的协作能力。
OpenFold3 现已发布,并可通过 NVIDIA NIM 获得额外加速。本文将介绍如何在结构预测任务中使用 OpenFold3 NIM。
快速入门链接
预备知识
- 从支持的操作系统开始安装 Docker
- 安装 Docker(建议版本:23.0.1 或更高)
- 安装 NVIDIA 驱动程序(建议版本:580,支持 CUDA 13.0)
- 安装 NVIDIA 容器工具包(建议版本:1.13.5 或更高)
- 查阅 OpenFold3 NIM 相关文档
使用 OpenFold3 NIM 进行结构预测
借助 OpenFold3 NIM,结构预测可轻松实现从原型到生产的快速过渡,具体步骤如下。
第 1 步:访问模型
您可以通过 build.nvidia.com 获取 OpenFold3 NIM,支持在本地、集群或作为托管 NIM 服务部署容器。
docker pull nvcr.io/nim/openfold/openfold3:latest
export LOCAL_NIM_CACHE=~/.cache/nim
export NGC_API_KEY=<Your NGC API Key>
docker run --rm --name openfold3 \
--runtime=nvidia \
--gpus 'device=0' \
-p 8000:8000 \
-e NGC_API_KEY \
-v $LOCAL_NIM_CACHE:/opt/nim/.cache \
--shm-size=16g \
nvcr.io/nim/openfold/openfold3:latest
第 2 步:提交结构预测作业
部署完成后,您可以通过标准的 REST 调用或 Python 客户端与 API 进行交互。
#!/usr/bin/env python3
import requests
import os
import json
from pathlib import Path
# Define output file and inference endpoint
output_file = "output.json"
url = "http://localhost:8000/biology/openfold/openfold3/predict"
# Define protein sequence
protein_sequence = "MGREEPLNHVEAERQRREKLNQRFYALRAVVPNVSKMDKASLLGDAIAYINELKSKVVKTESEKLQIKNQLEEVKLELAGRLEHHHHHH"
# Define MSA alignment in CSV format
msa_alignment_csv = "key,sequence\n-1,MGREEPLNHVEAERQRREKLNQRFYALRAVVPNVSKMDKASLLGDAIAYINELKSKVVKTESEKLQIKNQLEEVKLELAGRLEHHHHHH"
# Define DNA sequences (complementary pair)
dna_sequence_b = "AGGAACACGTGACCC"
dna_sequence_c = "TGGGTCACGTGTTCC"
# Build request data
data = {
"request_id": "5GNJ",
"inputs": [
{
"input_id": "5GNJ",
"molecules": [
{
"type": "protein",
"id": "A",
"sequence": protein_sequence,
"msa": {
"main_db": {
"csv": {
"alignment": msa_alignment_csv,
"format": "csv",
}
}
}
},
{
"type": "dna",
"id": "B",
"sequence": dna_sequence_b
},
{
"type": "dna",
"id": "C",
"sequence": dna_sequence_c
}
],
"output_format": "pdb"
}
]
}
r = requests.post(url=url, json=data)
# Save the json output
print(r, "Saving to output.json:\n", r.text[:200], "...")
Path(output_file).write_text(r.text)
预测包含3D坐标(PDB/mmCIF格式)以及置信度指标(如pLDDT、pTM和ipTM),所有预测均可在配备NVIDIA H100 Tensor Core GPU的设备上于数秒内完成。
蛋白质结构预测的新开放标准
OpenFold 联盟 是由 Bayer、Bristol Myers Squibb、Johnson& Johnson、Novo Nordisk、Outpace Bio 等公司组成的行业领导联盟,在推进开放、可复制的建模系统方面发挥了重要作用。
OpenFold3 是该联盟至今最重要的里程碑之一。该模型将结构预测能力拓展至多聚体、蛋白质-DNA/RNA复合物以及配体复合物,其预测精度达到甚至超越了当前领先的开源模型。
值得注意的是,OpenFold3 在蛋白质-核酸相互作用的基准测试中表现优异,性能已达到与 AlphaFold3 相当的水平,而此前的模型在这一领域普遍表现不足。此外,该系统被纳入 Linux 基金会开放模型定义中的 1 类开源项目,确保了其完全的透明度和可复现性。
开放科学符合企业可靠性
OpenFold3 已针对 NVIDIA 加速 AI 计算堆栈进行了优化,涵盖以下方面:
- cuEquivariance:面向 3D 对称运算的物理感知加速技术。
- MMseqs2-GPU:支持 GPU 原生的多序列比对工具,显著提升计算效率。
- NVIDIA FLARE:支持联邦学习,实现跨机构模型微调,无需共享数据即可协作。
这些集成使 OpenFold3 NIM 既便于开发者使用,也适合企业部署,可作为适用于本地、混合及云环境的即插即用服务。借助 NVIDIA TensorRT,大型多聚体和核酸复合物的推理速度可提升 1.8 倍。
OpenFold3 已通过 Apheris 与 SandboxAQ 在安全联合工作流中的验证,展现出其在国际制药研发环境中良好的可扩展性。借助该联合工作流,合作伙伴无需跨越机构边界传输数据,即可基于专有数据(如抗体-抗原复合物或RNA-配体组合)进行模型微调。
根据 Linux 基金会的开放模型定义,OpenFold3 属于第一类开放系统,使软件及联盟能够受益于快速发展的贡献者群体和基准生态系统,从而保障持续优化与长期可靠性。
通过集成 NVIDIA FLARE,各机构可在多个站点(如制药合作伙伴、研究联盟和医院)协同训练 OpenFold3,同时无需共享敏感数据。
该方法在支持监管合规性(如 GDPR 和 HIPAA)的同时,还能整合原本孤立的不同数据集,从而提升模型的性能。
打造开放蛋白质 AI 的未来
OpenFold3 不仅是一个模型,更奠定了未来十年蛋白质人工智能发展的基石。它凝聚了 OpenFold 联盟、开源科学、加速计算与联邦协作的成果,汇集全球 40 多家机构的智慧,确保这一工具既服务于全球科研人员,又满足企业级的可靠性与安全标准。
致谢
衷心感谢 OpenFold 联盟以及 SandboxAQ、Apheris 等合作伙伴在推动面向分子科学的开放加速 AI 方面所开展的协作。