생성형 AI는 특히 AlphaFold 및 RosettaFold와 같은 혁신적인 기술을 통해 신약 개발 방식과 생명공학 기업 및 연구 실험실에서 단백질 구조를 연구하는 방법을 변화시키고 있으며, 단백질 상호 작용에 대한 획기적인 인사이트를 제공합니다.
단백질은 역동적인 존재입니다. AlphaFold2 및 생물학/의학 분야 응용 사례에서 알 수 있듯이, 단백질의 고유 상태는 아미노산 서열로만 결정된다는 가설이 있습니다. 그러나 단일 단백질은 온도 변화와 같은 환경에 따라 보이는 여러 개의 뚜렷한 형태를 채택할 수 있습니다. 이러한 형태는 유연한 영역, 단백질 복합체 내의 상호 작용, 활성 상태와 비활성 상태 간의 전환을 반영할 수 있습니다.
스타트업을 위한 NVIDIA Inception 프로그램의 회원사인 Protai는 치료적으로 가장 적절하지 않을 수도 있는 단일 구조에 정착하는 대신, 서로 다른 단백질 상태 간의 구조 변화를 분석하여 특정 작용 기전(MOA)에 대한 가장 정확한 단백질 구조를 결정하는 것을 목표로 합니다.
Protai는 질량분석법 단백질체학(Mass Spectrometry Proteomics)과 AI를 활용하여 인류 건강과 사회에 실질적인 변화를 가져올 수 있는 정밀 의학 솔루션을 개발함으로써 신약 개발의 경계를 넓히고 있습니다. Protai 플랫폼의 핵심은 노벨상 수상에 빛나는 단백질 구조 알고리즘, 물리 기반 도구, 독점 단백질체학 데이터를 통합하는 단백질 구조 예측 파이프라인입니다.
Protai는 플랫폼에서 AI 추론 속도를 높이기 위해 신약 개발에 최적화된 생성형 AI 생물학 모델 세트인 NVIDIA NIM 마이크로서비스를 채택했습니다. NIM 마이크로서비스를 통해 Protai는 정확도를 손상시키지 않으면서 단백질 구조 예측을 위해 처리량을 획기적으로 늘리고 지연 시간을 단축했습니다. 이 게시물에서는 Protai가 어떻게 NVIDIA NIM을 통합하여 정확하고 확장 가능한 단백질 구조 예측을 지원하여 신약 개발 접근 방식을 변화시키는지 자세히 알아봅니다.
단백질 복합체 구조 예측 배경
단백질 복합체를 이해하는 것은 구조 생물학의 핵심적인 기반입니다. 단백질 복합체는 두 개 이상의 결합된 폴리펩티드 체인이 상호 작용하며 함께 기능하여 특정 생체학적 활동을 수행하는 집합체입니다. 다중 단백질이 상호 작용하여 형성되는 다중체 단백질은 중요한 생물학적 과정을 주도하며, 신약 개발에서 핵심 목표가 됩니다. 과학계는 실험적 또는 연산 기술을 기반으로 모든 단백질 단량체의 구조를 밝히는 데 있어 진전을 이루고 있지만, 단백질 복합체의 수는 기하급수적으로 더 증가했습니다. 따라서 이러한 복합체에 대한 구조 연구를 용이하게 하기 위한 예측 알고리즘의 필요성이 더욱 커지고 있습니다.
AlphaFold-Multimer는 다중체 단백질 구조의 고품질 컴퓨팅 예측을 가능하게 함으로써 이러한 한계를 극복합니다. AlphaFold를 기반으로 구축된 이 혁신은 딥 러닝을 사용하여 단백질 간 상호 작용을 해석합니다. AlphaFold 알고리즘, 매개변수, 출력, 배포에 대한 자세한 내용은 다음 섹션에서 제공할 예정입니다.
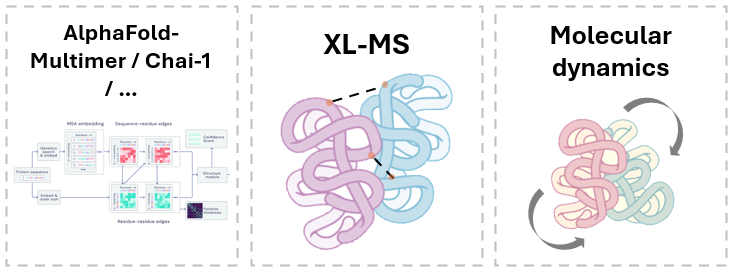
Protai는 다각적인 접근 방식을 취하고 있습니다. 한 가지 주요 전략은 AlphaFold-Multimer와 같은 구조 예측 모델의 샘플링을 향상시키는 것입니다. 또한, Protai는 자체 교차 연결 질량 분석(XL-MS) 데이터를 생성하여 서로 다른 상태에서 특정 단백질 구조를 나타내는 연결체를 식별합니다.
XL-MS는 화학 교차 연결체를 사용하여 단백질 내부 또는 단백질 간에 특정 아미노산 잔기를 공유 결합시켜, 공간 근접성과 상호 작용 부위를 포착하는 강력한 실험 기술입니다. 이러한 교차 연결 영역은 유용한 거리 제약 조건을 제공하여, 단백질 형태와 상호 작용을 보다 정확하게 매핑할 수 있도록 합니다. 실험을 통해 유도된 이러한 제약 조건을 고급 샘플링 기법 및 분자 동역학 시뮬레이션과 결합함으로써 연구원은 현재 공개 영역에서 제공되는 것 이상으로 단백질 구조를 생성할 수 있습니다.
사례 연구: H3-H4 단백질 복합체 예측
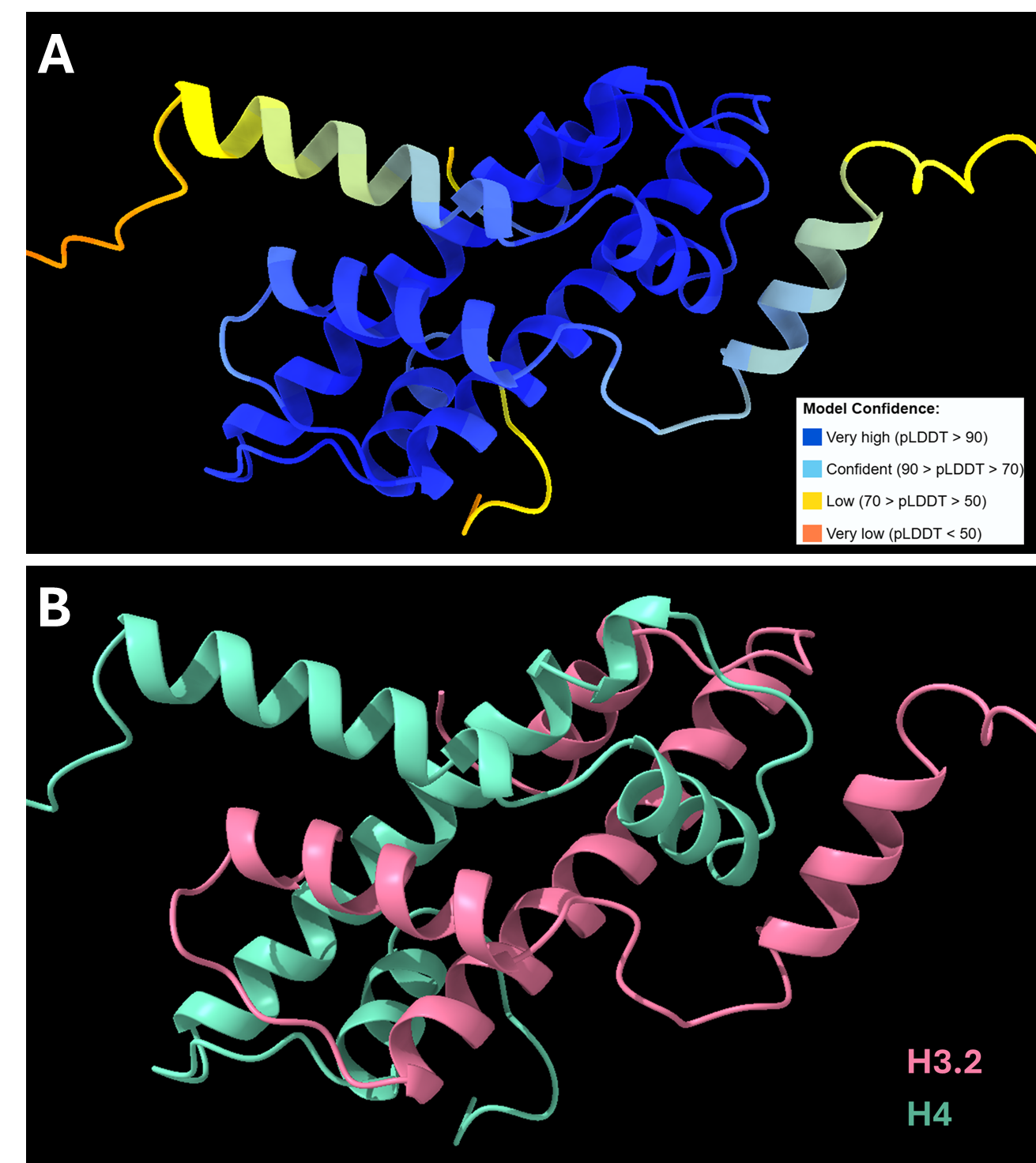
Protai의 역량을 보여주기 위해 이 섹션에서는 히스톤 H3 및 H4가 포함된 단백질 복합체의 예측을 살펴봅니다. 이 복합체는 전사 인자와 RNA 중합효소에 대한 DNA 접근성을 결정하는 데 핵심 역할을 하는 동시에, DNA 복구 과정 중에서 안정성을 유지하는 데에도 기여합니다.
Protai는 AlphaFold2-Multimer NIM을 사용하여 H3-H4 복합체의 구조를 예측했습니다. 결과로 생성된 구조는 신뢰도에 따라 색상 코드화되어 예측 정확도를 시각적으로 표현합니다. AlphaFold 훈련 세트에 포함된 단백질의 경우, 예측에 대한 신뢰도가 높은 경향이 있지만, 유연성과 고유한 특징은 특정 형태에 따라 달라질 수 있습니다.
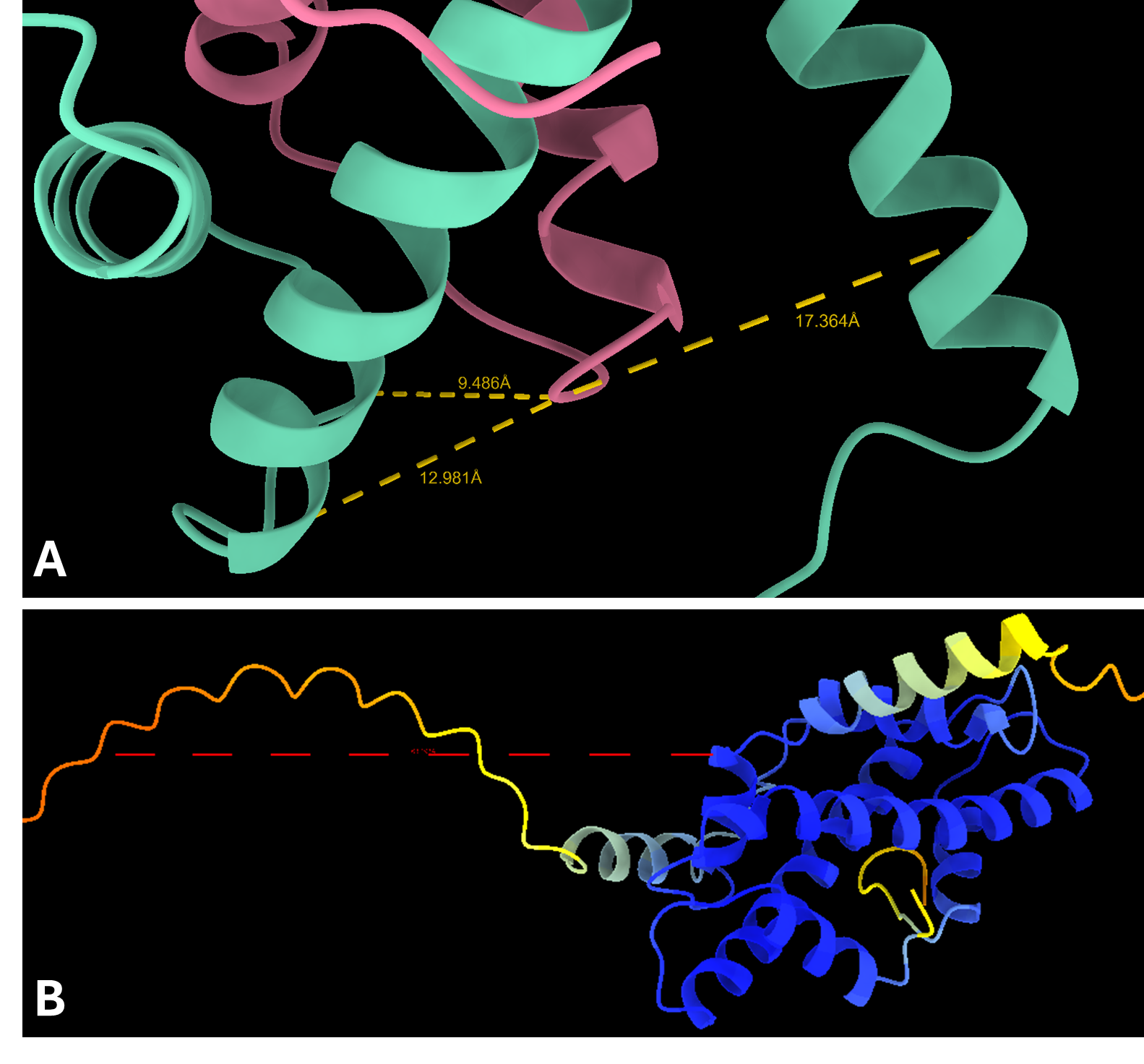
이러한 예측을 개선하기 위해 Protai는 XL-MS 데이터를 활용하여 H3와 H4 사이에 존재하는 3가지의 단백질 간 연결체를 식별했습니다. 이러한 연결체는 실험적 기준점 역할을 하며, Protai가 예측된 구조를 검증하거나 새로운 잠재적인 단백질 상태를 발견할 수 있도록 지원합니다. 이 사례에서는 상위 5개의 예측 결과 전반에서 3개의 연결체가 일관되게 유지되었습니다. 2개의 단백질에서 신뢰도가 높은 잔기와 낮은 잔기 사이에 추가 연결체가 확인되었으며, 이는 예측을 더욱 정밀하게 정의하고 향상시킬 수 있는 가능성을 보여줍니다.
NVIDIA NIM을 사용한 AlphaFold-Multimer 배포
AlphaFold-Multimer 및 기타 LLM 기반 도구의 확장 가능하고 최적화된 배포를 지원하기 위해 Protai는 NVIDIA NIM을 활용합니다. NIM은 NVIDIA GPU에서 원활한 배포를 위해 사전 최적화된 추론 컨테이너를 제공합니다. 이러한 컨테이너는 온프레미스 환경과 클라우드 환경 모두에서 최첨단 성능을 보장합니다. AlphaFold 자체는 단일 예측에 여러 개의 GPU를 활용할 수 없습니다. 그러나 NIM 마이크로서비스는 여러 개의 추론 작업을 병렬로 실행할 수 있도록 지원하여, 여러 단백질 복합체를 예측하는 데 필요한 전체 시간을 크게 단축할 수 있습니다. (대규모 복합체의 경우 예측에 24시간 이상 걸릴 수도 있습니다.)
워크플로우는 다음과 같은 주요 단계로 구성됩니다.
- 다중 서열 정렬(Multiple Sequence Alignment, MSA): MSA는 상호 작용 단백질 간의 보존 영역과 공동 진화 신호를 식별하여 예측을 위한 기반을 제공합니다. 기존 MSA 도구는 CPU 기반 구현에 의존하는데, 이는 순차 처리에는 효과적이지만 GPU 병렬 처리 능력에는 미치지 못합니다. AlphaFold2 NIM은 GPU 최적화된 서열 검색 및 클러스터링 제품군인 MMseqs2를 사용하며, 비교할 수 없는 속도로 효율적인 비교를 달성합니다.
- 단백질 상호 작용 모델링: AlphaFold-Multimer는 AlphaFold2 트랜스포머 기반 아키텍처의 수정된 버전을 사용합니다. 이는 단백질 복합체 구조 예측을 위해 체인 간 상호 작용에 맞게 미세 조정되었으며, 향상된 쌍 표현, 교차 체인 모델링, 다중체에 특화된 손실 함수를 사용합니다.
- 구조 개선: 예측된 구조는 정확성과 물리적 타당성을 보장하기 위해 정제 과정을 거치며, 입체 화학적 제약 조건과 실험적 벤치마크를 통합합니다.
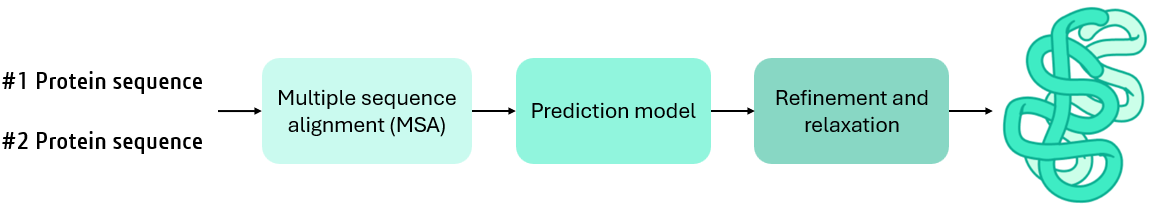
AlphaFold2-Multimer NIM은 추론 프로세스를 CPU 집약적인 작업과 GPU 집약적인 작업으로 분할할 수 있는 여러 엔드포인트를 제공하여 연산 비용을 절감합니다.
- protein-structure/alphafold2/multimer/predict-structure-from-sequences: 서열에서 전체 구조 예측(엔드 투 엔드)
- protein-structure/alphafold2/multimer/predict-MSA-from-sequences: 서열의 MSA 연산(CPU 집약적)
- protein-structure/alphafold2/multimer/predict-structure-from-MSA: 사전 연산된 MSA에서 구조 예측(GPU 집약적)
하드웨어의 선택은 단백질 복합체의 크기에 따라 달라집니다.
- 짧은 서열: 32GB의 GPU로 충분합니다.
- 더 큰 복합체(3,000개 이상의 잔기): 최적의 성능을 위해서는 NVIDIA H100 또는 A100 GPU가 필요합니다.
위의 사용 사례에서 Protai는 두 단백질이 작았기 때문에 NVIDIA L4 GPU에 AlphaFold2-Multimer NIM을 자체 배포했습니다.
매개변수
AlphaFold2-Multimer NIM은 다음 매개변수를 특징으로 합니다.
- sequences: 다중체 구조 예측이 수행될 표적 단백질 체인을 정의합니다.
- algorithm: 상동 서열을 검색하는 데 사용되는 알고리즘을 지정합니다. jackhmmer는 MSA 생성에 널리 사용되는 알고리즘이며, 모델이 단백질 간 상호 작용을 학습할 수 있도록 지원하는 진화적으로 관련된 서열을 식별합니다.
- e_value: 데이터베이스에서 상동 서열을 식별하기 위한 임계값으로, 값이 낮을수록 일치 기준이 더 엄격함을 나타냅니다.
- iterations: MSA 알고리즘이 수행하는 반복 횟수를 정의합니다(런타임 제한).
- databases: 정확한 MSA에 필요한 진화 정보를 쿼리하는 데이터베이스를 지정합니다.
- relax_prediction: 예측된 구조를 정제해야 하는지 여부를 나타냅니다. 결합 길이, 각도, 입체 화학적 제약 조건을 최적화하여 최종 구조의 물리적 타당성을 보장합니다.
출력
단백질 데이터 뱅크(PDB) 형식의 AlphaFold2-Multimer NIM 출력에는 예측된 단백질 다중체에 대한 원자 수준의 구조 정보가 포함되어 있습니다. 출력 파일에서 단백질의 각 원자는 PDB 형식 사양을 준수하는 구조화된 형식을 사용하여 기술됩니다.
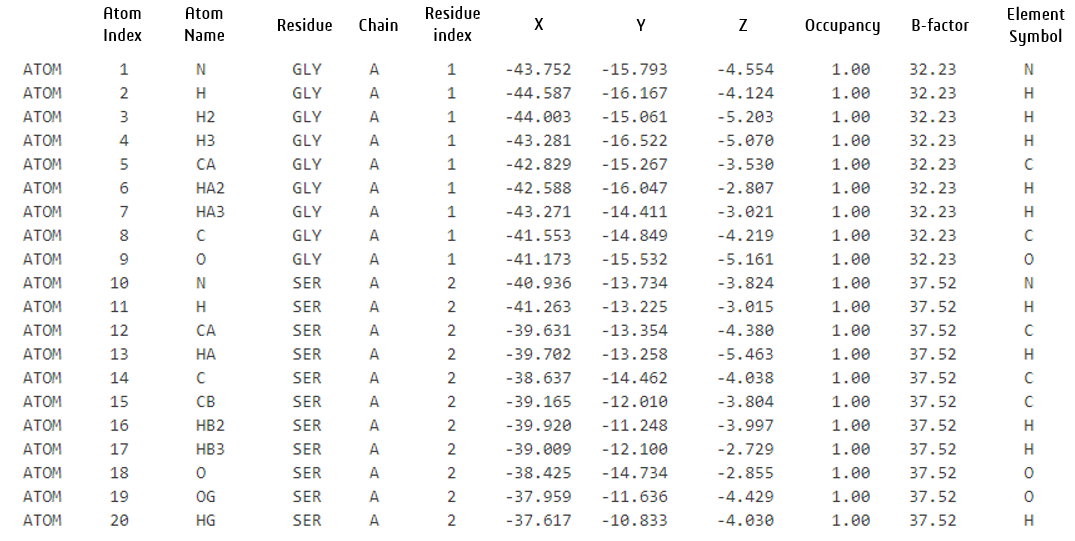
AlphaFold2-Multimer NIM을 사용하여 제공된 PDB 라인에 대한 설명은 다음과 같습니다.
- 체인 식별자: 출력에서 단백질 체인을 식별하는 데 사용됩니다. 예를 들어 두 개의 단백질로 구성된 단백질 복합체에는 최소 두 개의 체인이 있습니다.
- 좌표(X, Y, Z): 원자의 3D 공간 좌표(옹스트롬 Å 단위).
- 점유율: 이 위치에서 해당 원자가 관찰될 확률이며, 0.00(완전히 없음)에서 1.00(항상 존재) 사이의 값을 가집니다. 실험 데이터에서 하나의 원자가 결정 구조에서 여러 개의 형태로 나타나면 점유율은 1.00 미만일 수 있습니다(예: 여러 개의 회전 이성질체(대체 형태)를 가진 측쇄의 경우, 모든 형태에 대한 점유율을 합하면 1.00이 될 수 있습니다). AlphaFold 예측은 단백질에 대해 가장 가능성이 높은 단일 구조를 제공하기 때문에 모든 원자의 점유율은 1로 설정됩니다.
- B-factor: 구조 내에서 원자 변위 또는 유연성의 척도이며, 값이 클수록 움직임이 많음을 나타냅니다. 실험 데이터에서는 열 운동이나 무질서를 반영할 수 있습니다. AlphaFold 출력에서 B-factor는 예측된 로컬 거리 차이 테스트(Local Distance Difference Test, pLDDT)에서 파생된 신뢰도 점수로 대체됩니다. pLDDT는 0에서 100 사이의 값을 가지며, AlphaFold가 제공하는 잔기별 신뢰도 점수입니다.
결론
Protai의 구조 예측 워크플로우는 AlphaFold2-Multimer NIM과 실험을 통해 확인된 고유한 XL-MS 연결체와 결합합니다. Protai는 NVIDIA에 최적화된 AI 인프라를 활용하여 예측을 가속화하고 확장성을 향상시켰습니다. 이를 통해 이전에는 연구가 불가능했던 단백질 상호 작용을 탐색하여 신약 개발 및 정밀 의학 분야에서 새로운 가능성을 열 수 있습니다.
H3-H4 테스트 사례는 구조 예측과 XL-MS 데이터를 병합하는 Protai의 통합적 접근 방식이 어떻게 생물학적 기능과 치료 응용 분야를 이해하는 데 중요한 단백질 형태 및 동역학에 대한 인사이트를 제공하는지 보여줍니다.
생성형 AI가 계속 진화함에 따라, NVIDIA NIM 및 NVIDIA BioNeMo 프레임워크와 같은 도구는 연산 생물학의 잠재력을 최대한 활용하는 데 중요한 역할을 할 것입니다. Protai는 최첨단 AI 기술을 지속적으로 통합하며, 생명을 구하는 신약이 그 어느 때보다 빠르고 효율적이며 정밀하게 개발되는 미래에 더 가까이 다가갈 수 있도록 최선을 다하고 있습니다.
생성형 단백질 결합제 설계를 위한 NVIDIA BioNeMo Blueprint, 생성형 가상 스크리닝을 위한 NVIDIA BioNeMo Blueprint등 신약 개발에 필요한 NIM 마이크로서비스의 전체 세트를 실험할 수 있습니다. 또한 오픈 소스 BioNeMo 프레임워크로 자체 생물학 모델을 훈련함으로써 한 걸음 더 나아갈 수 있습니다. 마지막으로 GPU 가속 MMseqs2로 AlphaFold2 단백질 구조 예측 성능을 향상시키는 방법을 알아보세요.
관련 리소스
- GTC 세션: 농업 생명공학 형질을 위해 BioNemo를 사용하는 단백질 AI 응용 분야
- GTC 세션: NVIDIA BioNeMo NIM과 Blueprint로 신약 개발을 간소화
- GTC 세션: NVIDIA의 BioNemo, OpenFold, EquiDock 프레임워크를 사용하여 몇 초 안에 2억 개의 단백질-단백질 상호 작용 분석
- NGC 컨테이너: AlphaFold2-Multimer
- NGC 컨테이너: AlphaFold2
- SDK: BioNeMo 프레임워크