
A new paper published in Cell systems by Harvard researcher Mohammed AlQuraishi describes a new deep learning-based approach for predicting the 3D structure of a protein based on its amino acid sequence. The work achieves high accuracy at speeds upward of a million times faster than previous methods.
“Protein folding has been one of the most important problems for biochemists over the last half-century, and this approach represents a fundamentally new way of tackling that challenge,” said AlQuraishi. “We now have a whole new vista from which to explore protein folding, and I think we’ve just begun to scratch the surface.”
Using NVIDIA TITAN Xp GPUs with the cuDNN-accelerated TensorFlow deep learning framework, AlQuaraishi trained a recurrent geometric network on thousands of known proteins, with the model learning and improving its accuracy on every epoch.
Once trained, AlQuaraishi compared the performance of his model against other methods developed in recent years. His model outperformed all other previous methods in predicting protein structures and did so six to seven orders of magnitude faster than existing computational methods.
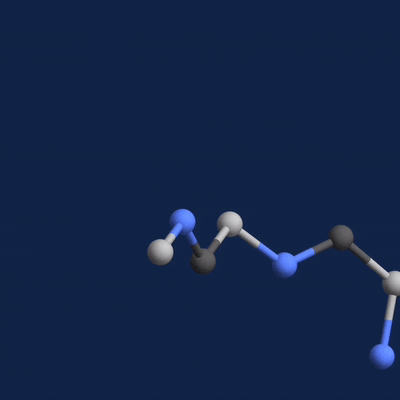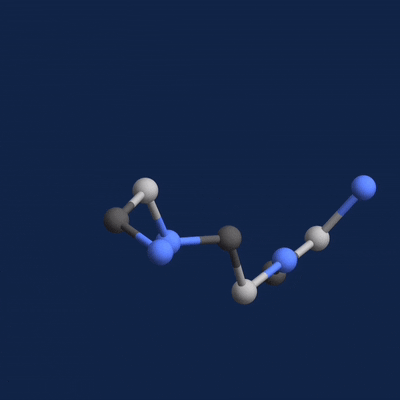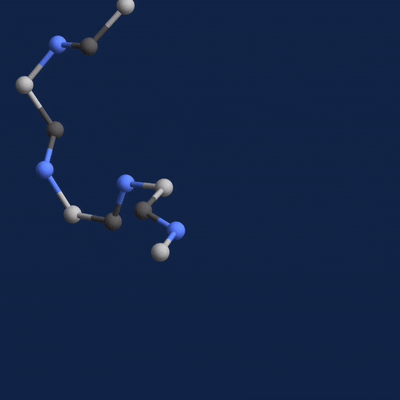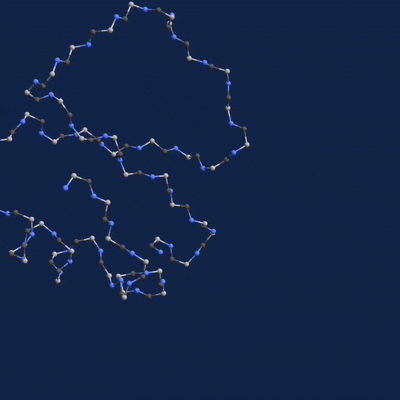
The GPUs were used for both training and inference, AlQuaraishi says.
“Accurately and efficiently predicting protein folding has been a holy grail for the field … We might solve this soon, and I think no one would have said that five years ago. It’s very exciting and also kind of shocking at the same time,” said AlQuaraishi.
Mohammed AlQuraishi presented his work on protein folding at a seminar at the Broad Institute of MIT and Harvard in March.
The new model is still in development and not ready for use in drug discovery or design, however, the software and results are now freely available on GitHub for developers and scientists to try.
