
分子动力学(MD)模拟是计算化学与材料科学中的重要工具,在微观层面研究化学反应、材料性质以及生物相互作用方面发挥着关键作用。然而,由于其高度的复杂性和巨大的计算需求,通常需要借助机器学习原子间势能模型(MLIP)等先进技术,以实现更好的可扩展性、计算效率和预测精度。
通过 ML-IAP-Kokkos 接口,可将基于 PyTorch 的机器学习势函数(MLIP)高效集成至 LAMMPS 分子动力学软件中,从而实现对化学与材料科学中原子系统的快速且可扩展的模拟。
该接口由 NVIDIA 联合洛斯阿拉莫斯国家实验室与桑迪亚国家实验室的科学家共同开发,旨在简化外部开发者与社区模型之间的交互,实现 MLIP 与 LAMMPS 的无缝集成,从而支持可扩展的分子动力学(MD)仿真。
ML-IAP-Kokkos 接口支持消息传递型 MLIP 模型,借助 LAMMPS 内置的通信功能,实现 GPU 间高效的数据传输,对利用多 GPU 计算能力开展大规模模拟具有重要意义。该接口通过 Cython 实现 Python 与基于 C++/Kokkos 的 LAMMPS 之间的桥接,确保仿真流程的端到端 GPU 加速,从而全面提升整体计算性能。
我们将逐步介绍如何连接您自定义的 PyTorch 模型,以实现可扩展的 LAMMPS 仿真。现在就开始吧。
准备所需的工具和环境
- 具备 LAMMPS 或其他分子动力学(MD)仿真工具的使用经验;
- 熟练掌握 Python,熟悉 PyTorch 及机器学习模型,有 Cython 经验者优先;
- 具备 cuEquivariance 支持的模型开发经验或 NVIDIA GPU 加速优化经验者更佳。
构建您自己的 ML-IAP-Kokkos 接口
第一步:配置环境
请确保已安装所有必需的软件。
- 基于 Kokkos、MPI 和 ML-IAP 构建的 LAMMPS
- 支持 PyTorch 的 Python 接口
- 包含已训练的 PyTorch MLIP 模型(可选支持 cuEquivariance)
第 2 步:安装支持 ML-IAP-Kokkos 和 Python 的 LAMMPS 版本
下载并安装支持 Kokkos、MPI、ML-IAP 和 Python 的 LAMMPS 版本,要求为 2025 年 9 月 或之后发布的版本。
安装支持 Python 的 LAMMPS 时,会同时在您的 Python 环境中安装 lammps 模块。
为方便使用,我们提供了一个预装并已编译 LAMMPS 的容器。
第 3 步:为首选模型开发 ML-IAP-Kokkos 接口
使用 ML-IAP-Kokkos 接口时,LAMMPS 在仿真过程中会调用 Python 解释器,执行您编写的原生 Python 模型,并支持完整的 Python 功能,而无需对模型进行编译。为此,需要确保运行环境中存在一个可访问模型类的有效 Python 环境。要将模型与 LAMMPS 集成,您需实现 `mliap_unified_abc.py` 文件中定义的 LAMMPS 抽象基类 MLIAPUnified。具体而言,需在 `compute_forces` 函数中,利用从 LAMMPS 传递过来的数据,完成能量和成对力的计算。该类还要求实现 compute_gradients 和 compute_descriptors 函数,但在这套配置中它们不会被调用,因此可留空。
我们首先通过一个简单的示例,了解 LAMMPS 传递给您的类的数据,然后创建一个能够输出相关信息的类。
from lammps.mliap.mliap_unified_abc import MLIAPUnified
import torch
class MLIAPMod(MLIAPUnified):
def __init__(
self,
element_types = None,
):
super().__init__()
self.ndescriptors = 1
self.element_types = element_types
self.rcutfac = 1.0 # Half of radial cutoff
self.nparams = 1
def compute_forces(self, data):
print(f"Total atoms: {data.ntotal}, Local atoms: {data.nlocal}")
print(f"Atom indices: {data.iatoms}, Atom types: {data.elems}")
print(f"Neighbor pairs: {data.npairs}")
print(f"Pair indices and displacement vectors: ")
print("\n".join([f" ({i}, {j}), {r}" for i,j,r in zip(data.pair_i, data.pair_j, data.rij)]))
请注意, __init__ 函数用于指定模型可处理的元素类型以及近邻列表的截断半径(减半),这些参数是模型设置中 LAMMPS 所必需的。
使用 PyTorch 的保存函数来存储模型对象。
mymodel = MLIAPMod(["H", "C", "O"])
torch.save(mymodel, "my_model.pt")
这将生成一个名为 file my_model.pt 的“pickled”Python 对象,LAMPS 可以加载该对象。在实际应用中,此类文件通常包含模型的权重,但此处仅是对类的序列化。由于类中可能包含任意 Python 代码,出于安全考虑,新版 PyTorch 默认禁止加载此类对象。通过设置环境变量 TORCH_FORCE_NO_WEIGHTS_ONLY_LOAD=1,可以启用类的加载功能,但请务必仅使用受信任的 *.pt 文件。
为进行测试,我们将在 sample.pos 文件中定义一个 CO 2 分子体系。
#The mandatory first line is a comment and skipped!
3 atoms
2 atom types
0.0 100.0 xlo xhi
0.0 100.0 ylo yhi
0.0 100.0 zlo zhi
Masses
1 12.0
2 16.0
Atoms
1 2 1.0 10.0 10.0
2 1 2.0 10.0 10.0
3 1 -0.1 10.0 10.0
该分子由一个碳原子和两个沿 X 轴线性排列的氧原子组成,碳原子位于 x = 1.0,两个氧原子分别位于 x = -0.1 和 x = 2.0。这一简化构型有助于清晰地观察 LAMMPS 如何处理轻原子与重原子之间的相互作用、成对作用力以及周期性边界条件,这些因素均为理解模拟系统中数据流动机制的关键。
图1展示了原子位置及其相对于模拟单元边界的关系。
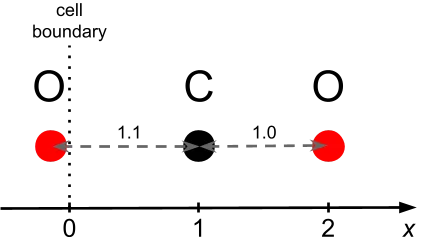
以下 LAMMPS 输入脚本 sample.in 用于加载原子坐标文件、设置相互作用势,并执行一次 MD 模拟步骤。
units metal
atom_style atomic
atom_modify map array
boundary p p p
read_data sample.pos
# Load the ML-IAP model
pair_style mliap unified my_model.pt 0
pair_coeff * * C O
dump dump0 all custom 1 output.xyz id type x y z fx fy fz
run 0
使用成对样式 mliap,关键字 “unified” 会指示 LAMMPS 启用 Python 接口,随后指定需加载的模型文件,最后的数值 0 用于控制近邻列表的设置(具体细节请参考 相关文档)。
我们选定感兴趣的元素子集(C 和 O),并指定它们在位置文件中的对应索引。
我们使用以下命令在单个 GPU 上运行 LAMMPS 与 Kokkos:
lmp -k on g 1 -sf kk -pk kokkos newton on neigh half -in sample.in
并查看输出中的相关部分:
Total atoms: 6, Local atoms: 3
Atom indices: [0 1 2], Atom types: [2 1 1 2 1 1]
Neighbor pairs: 4
Pair indices and displacement vectors:
(0, 5), [-1.1 0. 0. ]
(0, 1), [1. 0. 0.]
(1, 0), [-1. 0. 0.]
(2, 3), [1.1 0. 0. ]
与类相比,我们对数据结构有更深入的了解。
- 原子可分为两类:物理原子和重原子。物理原子的范围从
[0, nlocal)开始,需进行更新;重原子则从[nlocal, ntotal)开始,涵盖其他处理器或 GPU 上的数据,这些数据要么通过定期复制获得,要么来源于物理原子。 - 系统中共包含三个局部原子(
nlocal)和三个伪原子,因此原子总数为六个(ntotal)。 iatoms记录了局部原子的索引(即前 nlocal 个原子)。elems提供了所有原子的原子类型信息。npairs表示在截断范围内至少包含一个局部原子的原子对数量。需要注意的是,(i, j) 和 (j, i) 被视为两个独立的条目;例如,此处列出的四个原子对对应于 (C, O) 键在两个方向上的重复计数。
我们可以通过 pair_i、pair_j 和 rij 获取每对原子的 (i, j) 索引及其相对位移。需要注意的是,索引值大于或等于 nlocal 时,对应的是伪原子(例如,索引 3 表示周期性镜像的索引为 0,索引 5 表示周期性镜像的索引为 2)。
设想一个模型通过局部原子计算某些“节点特征”,并基于邻近原子的特征和键长来更新能量(这是机器学习势函数中的典型设置)。为简化起见,将所有特征值设为1,径向函数设为原子间距(在实际实现中,这些将由模型计算得到更为复杂的表达形式)。
实现这一潜力的部分可能包括:
def compute_forces(self, data):
rij = torch.as_tensor(data.rij)
rij.requires_grad_()
features = torch.zeros((data.ntotal,1)).cuda()
for i in range(data.nlocal):
features[i,0] = 1.0
Ei = torch.zeros(data.nlocal).cuda()
# Message passing step (j → i)
for r,i,j in zip(rij, torch.as_tensor(data.pair_i), torch.as_tensor(data.pair_j)):
Ei[i] += features[j,0] * torch.norm(r)
# Compute total energy and forces
Etotal = torch.sum(Ei)
Fij = torch.autograd.grad(Etotal, rij)[0]
# Copying total energy and forces
data.energy = Etotal
data.update_pair_forces_gpu(Fij)
请留意以下代码中的一些细节:
- 在 ML-IAP 与 LAMMPS 之间传递的所有物理量均采用双精度浮点数(fp64)并在设备端进行处理。模型内部允许类型转换以及主机到设备的数据迁移。
- 总能量在模型中被直接更新。
- 我们通过相对于 rij 的自动微分函数计算原子间作用力,即采用“每键”方式而非“每原子”方式计算力。因此,任何直接计算原子力的传统势函数都需转换为此种形式。
我们通过 update_pair_forces_gpu 函数更新 LAMMPS 中的力,并传入一个形状为 npairs, 3 的 fp64 类型 GPU 张量。
我们在 CO 2 这个小分子上测试了该代码。
作为参考,我们仅使用两对原子及两种距离(1 和 1.1)手动计算能量和作用力。总能量应为 4.2,每对原子间的作用力为 1;碳原子受到方向相反的两个力,合力为 2,而氧原子受力为 0。
然而,在运行此代码时,我们得到的总能量为 2(如 stdout 所示),且作用力也仅出现在一个碳原子和一个氧原子上(如 output.xyz 所示)。显然,问题出在伪原子的存在:它们的贡献并未正确传递到真实原子上,因此只有 (0, 1) 键对总能量和作用力有贡献。
在这种简化的设定下,我们可以将重物初始化为 1,但在实际场景中,这可能涉及更复杂的环境函数。为此,我们利用 ML-IAP-Kokkos 中内置的消息传递机制,将重影特征更新为其对应真实粒子的值。该方法能够有效处理由周期性边界条件产生的重粒子,以及分布在不同 GPU 上的重粒子。
为协助该初始化过程,我们提供了两个例程:forward_exchange 用于在物理对应对象中设置伪原子,reverse_exchange 用于将伪原子的贡献汇总至物理对应对象。
第 4 步:为 ML-IAP-Kokkos 实现消息传递支持
为更新模型以采用消息传递机制(参见下方代码),我们将调用 `forward_exchange` 函数。该函数接收两个形状相同的张量 ntotal, feat_size,并将 features[0,nlocal) 设置为 new_features[0,ntotal),以统一处理本地数据复制和 MPI 消息传递操作。其中,feat_size (1) 表示特征向量的宽度。该函数会利用物理对应关系所定义的重影区域值,创建特征的副本。
def compute_forces(self, data):
rij = torch.as_tensor(data.rij)
rij.requires_grad_()
features = torch.zeros((data.ntotal,1)).cuda()
for i in range(data.nlocal):
features[i,0] = 1.0
Ei = torch.zeros(data.nlocal).cuda()
# Update of the features
new_features = torch.empty_like(features)
data.forward_exchange(features, new_features, 1)
features = new_features
# Local message passing step
for r,i,j in zip(rij, torch.as_tensor(data.pair_i), torch.as_tensor(data.pair_j)):
Ei[i] += features[j,0] * torch.norm(r)
# Compute total energy and forces
Etotal = torch.sum(Ei)
Fij = torch.autograd.grad(Etotal, rij)[0]
# Copying total energy and forces
data.energy = Etotal
data.update_pair_forces_gpu(Fij)
再次运行该代码后,我们得到了正确的能量和作用力。然而,在使用多个 GPU 时,力的计算无法正常工作,因为梯度并未在不同设备之间进行跨设备计算。
为解决该问题,我们需要使用 PyTorch 定义并注册 LAMMPS_MP.forward() 的梯度,而 forward_exchange 的梯度则由 reverse_exchange 提供。在调用 r everse_exchange 后,梯度将仅保留于真实原子,而在所有伪原子上的梯度将被置零。
以下是一个简单的消息传递实现:
import torch
class LAMMPS_MP(torch.autograd.Function):
@staticmethod
def forward(ctx, *args):
feats, data = args # unpack
ctx.vec_len = feats.shape[-1]
ctx.data = data
out = torch.empty_like(feats)
data.forward_exchange(feats, out, ctx.vec_len)
return out
@staticmethod
def backward(ctx, *grad_outputs):
(grad,) = grad_outputs # unpack
gout = torch.empty_like(grad)
ctx.data.reverse_exchange(grad, gout, ctx.vec_len)
return gout, None
有了这个新类,插件的最终形态将调整为:
def compute_forces(self, data):
rij = torch.as_tensor(data.rij)
rij.requires_grad_()
features = torch.zeros((data.ntotal,1)).cuda()
for i in range(data.nlocal):
features[i,0] = 1.0
Ei = torch.zeros(data.nlocal).cuda()
# Update of the features
features = LAMMPS_MP().apply(features, data)
# Local message passing step
for r,i,j in zip(rij, torch.as_tensor(data.pair_i), torch.as_tensor(data.pair_j)):
Ei[i] += features[j,0] * torch.norm(r)
# Compute total energy and forces
Etotal = torch.sum(Ei)
Fij = torch.autograd.grad(Etotal, rij)[0]
# Copying total energy and forces
data.energy = Etotal
data.update_pair_forces_gpu(Fij)
我们现在可以访问支持自动梯度计算的多 GPU 消息传递功能。
该结构应独立于消息传递的机器学习模型运行。
在传递任何消息之前调用 LAMMPS_MP 可确保特征保持同步,从而使模型正常运行。然而,在某些优化场景下,部分通信步骤可以被省略。由于 LAMMPS 在调用 ML-IAP 之前会收集重影原子信息,并在之后将作用于重影原子的力回传至其他计算单元,因此在多层 MLIP 中,通常可以省略首次的正向通信和末次的反向通信。
如前所述,力的计算需要求解键能的梯度。此外,通过将所有相关项传递给 LAMMPS,可由 LAMMPS 自行计算应力的贡献,插件本身无需计算应力。
使用 HIPPYNN 模型评估性能表现
为评估消息传递 Hook 带来的性能提升,我们对基于 HIPPYNN(Chigaev 2023)模型的 LAMMPS 进行了基准测试,实验在 1 至 512 个 NVIDIA H100 GPU 上运行。测试涵盖了包含 h = 1 到 h = 4 个交互层的机器学习原子间势(MLIP)模型,并对比了启用与未启用通信 Hook 的情况。性能测试采用简单的铝原子晶格,每个 GPU 上维持约 203 个原子,以确保负载均衡。
这些模型基于论文中的数据集进行训练,训练过程参考了 HIPPYNN 中的多交互层示例脚本,用于自动构建铝体系的稳健原子间相互作用势。为确保仿真的准确性,其中 r 表示 MLIAP 层的相互作用半径;在启用 hook 时,采用带 hook 功能的重影截断长度 r+epsilon,而在禁用 hook 时,则在近邻列表中包含 Ghost-Ghost 相互作用对,对应参数为 hr+epsilon。
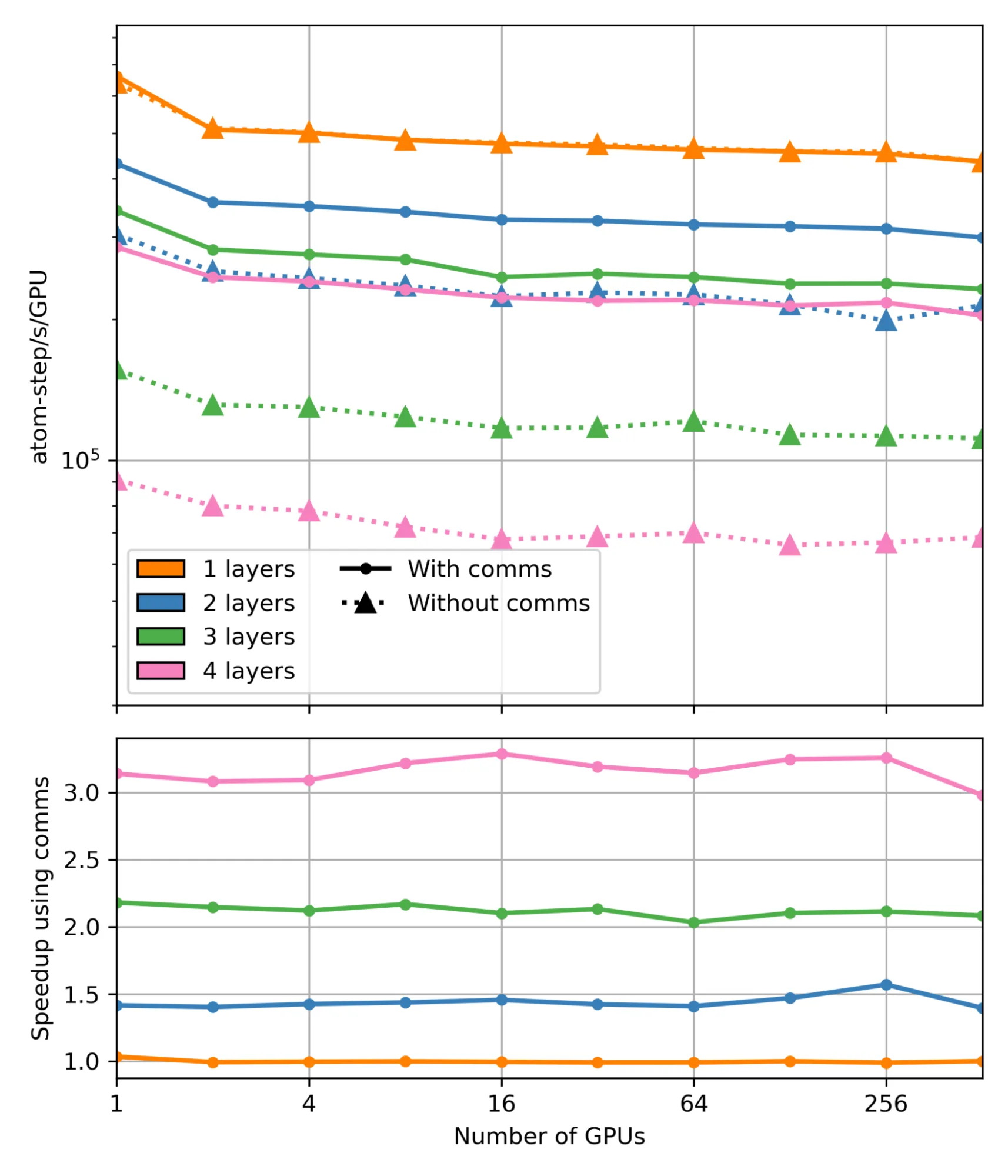
通过使用上图中的通信 hook 实现加速的主要原因是减少了伪原子的数量。在未启用 hook 时,包含一层、两层、三层和四层幽灵原子的结构中,真实原子占总原子数(真实原子与幽灵原子之和)的比例分别为 54%、38%、26% 和 18%。启用 hook 后,只需使用一个半径的重影原子,因此在所有情况下真实原子的比例均可保持在 54%。这一优化使得两层、三层和四层结构的总原子数分别减少了约 1.4 倍、2.1 倍和 3 倍(对于一层结构,hook 不起作用)。原子总数的减少与所观察到的计算加速直接对应。
使用 MACE 进行集成对比分析
为进一步演示,我们采用流行的机器学习势函数(MLIP)MACE 对 LAMMPS 模拟进行基准测试,比较了两种实现方式:一种是 LAMMPS 中原始的 MACE 配对样式,通过 C++ 中的 libtorch 加载模型权重;另一种是基于 cuEquivariance 的 MACE 插件,通过 MLIAP-Kokkos 接口实现。其中,cuEquivariance 是 NVIDIA 开发的一个用于加速几何神经网络的 Python 库。
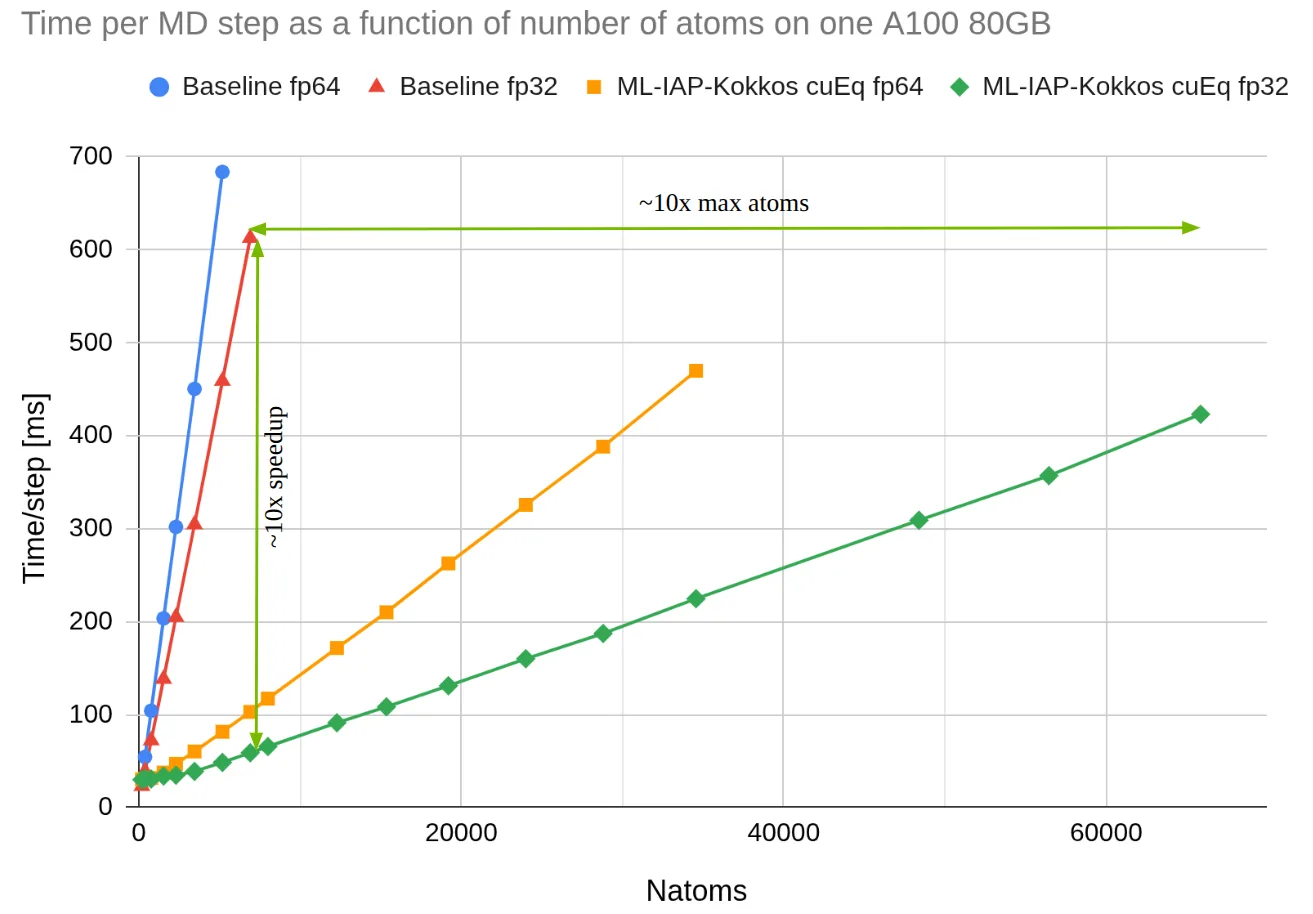
在本项基准测试中,我们使用 MACE-OFF23 电势的“中等”版本,在配备 80GB 显存的单个 A100 GPU 上模拟规模逐步增大的水箱系统,直至 GPU 显存耗尽。测试同时展示了原始 MACE 风格的性能作为对比。结果表明,新插件在计算速度和内存效率方面均有提升。性能提升主要得益于 cuEquivariance 对模型的加速,以及 MLIAP-Kokkos 接口中消息传递机制的优化。通过 ML-IAP-Kokkos 接口集成后,经 cuEquivariance 加速的机器学习势函数可无缝集成至 LAMMPS 中使用。
总结
LAMMPS ML-IAP-Kokkos 接口是实现基于机器学习势函数(MLIP)的多GPU、多节点分子动力学(MD)模拟的重要工具。它能够高效地模拟超大规模体系,有效弥合了现代机器学习力场与高性能计算基础设施之间的差距。
通过 ML-IAP-Kokkos 接口,可将基于 PyTorch 的机器学习势函数(MLIP)与 LAMMPS 有效集成,从而实现可扩展且高效的分子动力学(MD)模拟。本教程概述了配置和运行模拟所需的关键步骤,并重点介绍了 LAMMPS 在 GPU 加速和消息传递方面的优势。基准测试结果进一步展示了该接口在大规模模拟中的潜力,为相关领域的研究人员和开发者提供了一个强有力的工具。
准备好加速您的模拟体验了吗?立即尝试教程,并与社区分享您的成果。订阅 NVIDIA 新闻 并在 LinkedIn、X、Discord 和 YouTube 上关注 NVIDIA AI,获取最新动态。




