NVBIO is an open-source C++ template library of high performance parallel algorithms and containers designed by NVIDIA to accelerate sequence analysis and bioinformatics applications. NVBIO has a threefold focus:
- Performance, providing a suite of state-of-the-art parallel algorithms that offer a significant leap in performance;
- Reusability, providing a suite of highly expressive and flexible template algorithms that can be easily configured and adjusted to the many different usage scenarios typical in bioinformatics;
- Portability, providing a completely cross-platform suite of tools, that can be easily switched from running on NVIDIA GPUs to multi-core CPUs by changing a single template parameter.
Exponential Parallelism
We built NVBIO because we believe only the exponentially increasing parallelism of many-core GPU architectures can provide the immense computational capability required by the exponentially increasing sequencing throughput.
There is a common misconception that GPUs only excel at highly regular, floating point intensive applications, but today’s GPUs are fully programmable parallel processors, offering superior memory bandwidth and latency hiding characteristics, and R&D efforts at NVIDIA and elsewhere have proved that they can be a perfect match even for branchy, integer-heavy bioinformatics applications. The caveat is that legacy applications need to be rethought for fine-grained parallelism.
Many CPU algorithms are designed to run on few cores and scale to a tiny number of threads. When the number of threads is measured in the thousands, rather than dozens—a fact that all applications inevitably must consider—applications must tackle fundamental problems related to load balancing, synchronization, and execution and memory divergence.
NVBIO does just that, providing both low-level primitives that can be used from either CPU/host or GPU/device threads, as well as novel, highly parallel high-level primitives designed to scale from the ground up.
Broad Feature Spectrum
NVBIO offers a fairly broad suite of features; here is a short summary of some of the most relevant ones.
- Wide support for many string and string-set representations, including packed encodings for arbitrary alphabets (e.g. DNA, DNA+N, IUPAC, Protein, ASCII, etc).
- Parallel FM-index and Bidirectional FM-index construction and lookup for arbitrary string and string-sets.
- Novel, state-of-the-art parallel suffix sorting and BWT construction algorithms for very large strings and string sets, containing hundreds of billions of symbols. (Also see a paper on our parallel BWT algorithm.)
- Wavelet Trees and other succint data structures of large alphabet strings.
- Efficient parallel k-Maximal Extension Match filters
- Parallel Bloom Filter construction and lookup.
- State-of-the-art Q-Gram Index (also known as kmer index) construction and lookup.
- A state-of-the-art Dynamic Programming Alignment module, containing Edit-Distance, Smith-Waterman and Gotoh aligners with support for global, semi-global and local alignment in both banded and full matrices, on arbitrary string encodings.
- A high performance I/O module supporting common sequencing formats (e.g. FASTA, FASTQ, BAM) with transparent, parallel compression and decompression.
Expressive Performance Portability
Overall, the three guiding principles mentioned in the introduction could be probably summarized in a single phrase: expressive performance portability. Unlike other libraries which focus on solving a single specific instance of a problem on predetermined data types, NVBIO embraces the STL paradigm [as does Thrust] and offers fully configurable algorithms and containers, which are adapted and optimized at compile time for the required user-defined types and the backend architecture.
For example, consider the problem of finding the optimal alignment of two strings. Many existing libraries target different kinds of dynamic programming algorithms; for example, SSW for Smith-Waterman alignment of ASCII strings on the CPU, or CUDASW++ for the GPU. But most of these libraries tackle the solution of a single, specific problem, on a single platform, and are not very flexible.
However, in bioinformatics more than in other fields, developers use many variants of similar algorithms in specific situations. For example, one application might require use of the Gotoh Smith-Waterman scoring with affine gap penalties to compute the global aligment score of two ASCII encoded protein strings, while a short read mapper might want to use the fast Myers bit-vector algorithm for computing the edit-distance between two DNA strings packed with two bits per characters, while yet another application might need to perform banded, local alignment of four-bit packed DNA+N strings with the plain Smith-Waterman scoring.
Predetermining and hardwiring all possibilities would be essentially impossible: as the following table shows it would require 
| Aligner | Alignment Type | Matrix Type | Pattern Type | Text Type | Backend |
| Edit-Distance | Global | Full | ASCII | ASCII | CPU |
| Smith-Waterman | Local | Banded | DNA | DNA | GPU |
| Gotoh | Semi-Global | DNA+N | DNA+N |
NVBIO takes a different path, enabling the user to quickly select the most appropriate algorithms and containers through simple template specialization, as the following example code shows.
#include <nvbio/basic/packed_vector.h>
#include <nvbio/strings/alphabet.h>
#include <nvbio/alignment/alignment.h>
using namespace nvbio;
void alignment_test()
{
const char* text = "AAAAGGGTGCTCAA";
const char* pattern = "GGGTGCTCAA";
// our DNA alphabet size, in bits
const uint32 ALPHABET_SIZE = AlphabetTraits<DNA>::SYMBOL_SIZE;
// instantiate two DNA packed host vectors
PackedVector<host_tag, ALPHABET_SIZE> dna_text( length(text) );
PackedVector<host_tag, ALPHABET_SIZE> dna_pattern( length(pattern) );
// and fill it in with the contents of our ASCII strings, converted
// to a 2-bit DNA alphabet (i.e. A = 0, C = 1, G = 2, T = 3)
assign(length(text), from_string<DNA>(text), dna_text.begin());
assign(length(pattern), from_string<DNA>(pattern),
dna_pattern.begin());
// perform a banded edit-distance, semi-global alignment
const uint32 BAND_WIDTH = 5;
const int32 ed = aln::banded_alignment_score<BAND_WIDTH>(
// build an edit-distance aligner
aln::make_edit_distance_aligner<aln::SEMI_GLOBAL>(),
dna_pattern, // pattern string
dna_text, // text string
-255 ); // minimum accepted score
// build a Gotoh scoring scheme
const aln::SimpleGotohScheme scoring(
2, // match bonus
-1, // mismatch penalty
-1, // gap open penalty
-1 ) // gap extension penalty
// perform a full-matrix, local alignment
aln::Best2Sink<int32> best2; // keep the best 2 scores
aln::alignment_score(
// build a local Gotoh aligner
aln::make_gotoh_aligner<aln::LOCAL>(scoring),
dna_pattern, // pattern string
dna_text, // text string
-255, // minimum accepted score
best2 ); // alignment sink
}
Notice that while the alignment functions in this case have been invoked by a host thread on iterators over host containers, they can just as easily be invoked by a GPU thread inside a CUDA kernel!
NVBIO also offers parallel batch alignment functions, that can be used to easily align pairs of corresponding strings in two string sets, as the following example shows.
void batch_alignment_test()
{
// build two concatenated string sets, one for the patterns,
// containing a concatenated sequence of strings of 100
// characters each, and one for the texts,
// containing 200 characters each
const uint32 n_strings = 10000;
const uint32 pattern_len = 100;
const uint32 text_len = 200;
// setup the strings on the host
nvbio::vector<host_tag, uint8> h_pattern(n_strings * pattern_len);
nvbio::vector<host_tag, uint8> h_text(n_strings * text_len);
// fill their content with random characters
for (uint32 i = 0; i < h_pattern.size(); i++)
h_pattern[i] = uint8( drand48() * 255.0f );
for (uint32 i = 0; i < text_storage.size(); i++)
h_text[i] = uint8( drand48() * 255.0f );
// copy the strings storage to the device
nvbio::vector<device_tag, uint8> d_pattern( h_pattern );
nvbio::vector<device_tag, uint8> d_text( h_text );
// allocate two vectors representing the string offsets
nvbio::vector<device_tag, uint32> d_pattern_offsets( n_strings+1 );
nvbio::vector<device_tag, uint32> d_text_offsets( n_strings+1 );
// prepare the string offsets using Thrust's sequence()
// function, setting up the offset of pattern i as i * pattern_len,
// and the offset of text i as i * text_len
thrust::sequence( d_pattern_offsets.begin(),
d_pattern_offsets.end(), 0u, pattern_len );
thrust::sequence( d_text_offsets.begin(),
d_text_offsets.end(), 0u, text_len );
// prepare a vector of alignment sinks
nvbio::vector<device_tag, aln::BestSink<uint32> >
sinks( n_strings );
// and execute the batch alignment, on a GPU device
aln::batch_alignment_score(
aln::make_edit_distance_aligner
<aln::SEMI_GLOBAL, MyersTag<ALPHABET_SIZE> >(),
make_concatenated_string_set( n_strings,
d_pattern_storage.begin(),
d_pattern_offsets.begin() ),
make_concatenated_string_set( n_strings,
d_text_storage.begin(),
d_text_offsets.begin() ),
sinks.begin(),
aln::DeviceThreadScheduler() );
}
High Composability
NVBIO’s containers and algorithms are designed to be highly composable. For example, consider how many aligners use ad-hoc FM-index implementations to perform seed mapping on DNA strands. Now, what if after having spent several months working on your custom FM-index you’d want to write a protein search tool?
Proteins require a much larger alphabet, and the simple rank dictionaries based on occurrence count tables require exponential storage in the number of bits per character. NVBIO’s Wavelet Trees can be used as an efficient rank dictionary for large alphabet strings, and can be easily used together with NVBIO’s FM-index class, as shown in the following example (See the full example on Github).
void protein_search()
{
// this string will be our protein database we want to search
// just a short string with all protein characters in sequence
const char* proteins = "ACDEFGHIKLMNOPQRSTVWYBZX";
const uint32 text_len = 24;
const uint32 alphabet_bits =
AlphabetTraits< PROTEIN >::SYMBOL_SIZE;
const uint32 alphabet_size = 1u << alphabet_bits;
// allocate a host packed vector
PackedVector<host_tag, alphabet_bits, true> h_text( text_len );
// pack the string
from_string<PROTEIN>( proteins,
proteins + text_len,
h_text.begin() );
// copy it to the device
PackedVector<device_tag, alphabet_bits, true> d_text( h_text );
// allocate a vector for the BWT
PackedVector<device_tag, alphabet_bits, true> d_bwt( text_len + 1 );
// build the BWT
const uint32 primary = cuda::bwt( text_len,
d_text.begin(),
d_bwt.begin(), NULL );
// define our wavelet tree storage type and its plain view
typedef WaveletTreeStorage< device_tag > wavelet_tree_type;
typedef WaveletTreeStorage< device_tag >::const_plain_view_type
wavelet_tree_view_type;
// build a wavelet tree
wavelet_tree_type wavelet_bwt;
// setup the wavelet tree
setup( text_len, d_bwt.begin(), wavelet_bwt );
typedef nvbio::vector<device_tag, uint32>::const_iterator
l2_iterator;
typedef fm_index<wavelet_tree_view_type, null_type, l2_iterator>
fm_index_type;
// take the const view of the wavelet tree
const wavelet_tree_view_type wavelet_bwt_view =
plain_view( (const wavelet_tree_type&)wavelet_bwt );
// build the L2 table
nvbio::vector<device_tag,uint32> L2(alphabet_size+1);
// note we perform this on the host, even though the wavelet
// tree is on the device - gonna be slow, but it's not much
// stuff, and this is just an example anyway... and we just
// want to show that NVBIO is designed to make everything work!
L2[0] = 0;
for (uint32 i = 1; i <= alphabet_size; ++i)
L2[i] = L2[i-1] + rank( wavelet_bwt_view, text_len, i-1u );
// build the FM-index
const fm_index_type fmi(
text_len,
primary,
L2.begin(),
wavelet_bwt_view,
null_type() );
// do some string matching using our newly built FM-index - once
// again we are doing it on the host, though all data is on the
// device: the entire loop would be better moved to the device
// in a real app.
fprintf(stderr, " string matching:\n");
for (uint32 i = 0; i < text_len; ++i)
{
// match the i-th suffix of the text
const uint32 pattern_len = text_len - i;
// compute the SA range containing the occurrences
// of the pattern we are after
const uint2 range = match( fmi,
d_text.begin() + i,
pattern_len );
// print the number of occurrences of our pattern,
// equal to the SA range size
fprintf(stderr, " rank(%s): %u\n",
proteins + i, 1u + range.y - range.x);
}
}
High Performance
The following graphs show the staggering performance of some of NVBIO’s parallel algorithms running on NVIDIA Tesla K40 Accelerators, for tasks such as FM-index lookup, DP alignment and BWT construction for very large read collections.
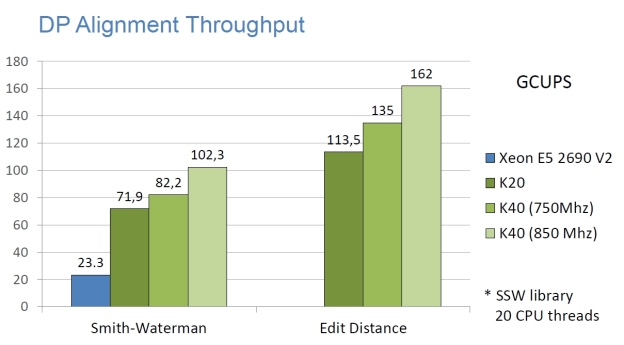
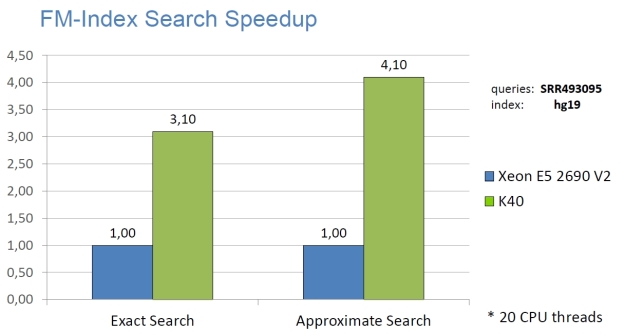
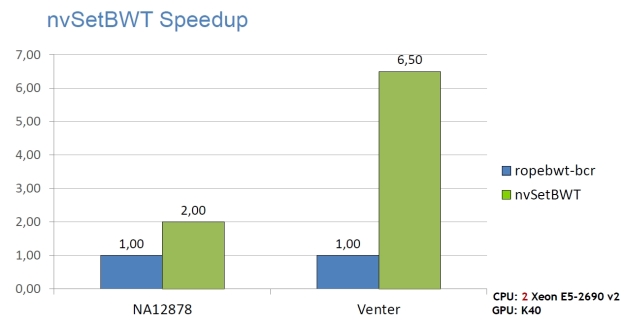
Interoperability
NVBIO is interoperable both with the high level Thrust C++ parallel algorithms library and lower level OpenMP and CUDA environments. Like Thrust, NVBIO offers full performance portability, providing both CUDA and multi-core CPU implementations for most of its algorithms.
Applications
NVBIO contains a suite of prebuilt high-performance applications.
- nvBowtie: an industrial-strength replacement for the popular bowtie2 aligner, providing almost the same exact specificity and sensitivity characteristics, at a great speed advantage.
- nvBWT: a single-string BWT construction application, which can construct the BWT of a human genome in just under 2 minutes on a single Tesla K40—compared to the 15+ minutes required by state-of-the-art CPU software.
- nvSetBWT: a state-of-the-art read indexing application, which can build the BWT of 1.2B x 100bp reads in under 1.7 hours.
Open Source
NVBIO is open-source software, distributed under the BSD Licence. The primary developers are Jacopo Pantaleoni and Nuno Subtil.
Try NVBIO Today
You can get NVBIO on Github at http://nvlabs.github.io/nvbio. NVBIO has been designed for GPUs supporting at least CUDA Compute Capability 3.5. Due to the high memory requirements typical of bionformatics algorithms, NVIDIA Tesla K20, K20x or K40 and above GPUs are recommended.
NVBIO is under active development, and as we are writing this article, we are targeting even more applications and bioinformatics domains. If you have specific comments or requests, please comment below or check out the NVBIO Users Forum.