GROMACS是一个用于生物分子系统的模拟软件包,是世界范围内使用率最高的科学软件应用程序之一,也是理解重要生物过程(包括当前新冠病毒大流行的基础)的关键工具。
在以前的职位中,我们展示了最近与核心开发团队合作进行的优化,这些优化使 GROMACS 在现代多 GPU 服务器上的运行速度比以前快得多。这些优化包括将计算和通信卸载到 GPU ,后者对于那些可以有效地将多个 GPU 并行用于单个模拟的相对较大的情况尤其有利。有关最新异构软件引擎的并行化和内部工作的更多信息,请参阅GROMACS 中分子动力学模拟的非均匀并行化和加速。
另一个越来越常见的工作流涉及运行许多独立的 GROMACS 仿真,其中每个仿真都可以相对较小。 NVIDIA GPU 的规模和能力不断增加,但通常情况下,相对较小的仿真系统的孤立单弹道仿真无法充分利用每个系统上的所有可用计算资源 GPU 。
但是,多部门工作流程可以涉及数十到数千个松散耦合的分子动力学模拟。在此类工作负载中,目标通常不是最小化单个仿真的求解时间,而是最大化整个集成的吞吐量。
按照 GPU 并行运行多个模拟可以显著提高总体吞吐量,正如之前在GROMACS 中分子动力学模拟的非均匀并行化和加速中针对 GROMACS 所示(图 11 )。已创建 NVIDIA 多进程服务器( MPS )和多实例 GPU ( MIG )功能,以促进此类工作流,通过使每个 GPU 能够同时用于多个任务,进一步提高效率。
在这篇文章中,我们演示了为 GROMACS 按照 GPU 运行多个模拟的好处。我们展示了如何利用 MPS ,包括与 MIG 结合使用,在每个 GPU 上并行运行多个模拟,从而实现高达 1 . 8 倍的吞吐量总体改善。
背景资料
硬件
对于本文后面给出的结果,我们在DGX A100服务器上运行,该服务器是 NVIDIA 内部赛琳娜超级计算机的一部分,它具有八个 A100-SXM4-80GB GPU 和两个 AMD EPYC 7742 (罗马) 64 核 CPU 插槽。
GROMACS 测试用例
为了生成下一节给出的结果,我们使用 24K 原子核糖核酸酶立方和 96K 原子 ADH-dodec 测试用例,这两个测试用例都使用 PME 进行远程电子 CTR 静态测试。有关更多信息和输入文件,请参阅GROMACS 中分子动力学模拟的异构并行化和加速补充信息。
我们使用 GROMACS 版本 2021 . 2 (与 CUDA 版本 11 . 2 、驱动程序版本 470 . 57 . 02 一起使用)。对于单个孤立实例(在单个A100-SXM4-80GB GPU 上),使用与后面所述相同的 GROMACS 选项,我们实现了 RNAse 的1083 纳秒/天和 ADH 的378 纳秒/天。
-update gpu选项对此处的性能至关重要。这将触发“ GPU – 驻留步骤”,其中每个 timestep 的更新和约束部分被卸载到 GPU 以及默认的“ GPU 强制卸载”行为。如果没有这一点,在这个硬件上,每种情况下的性能大约降低 2 倍。这对 CPU 和 GPU 可用功能的平衡非常敏感,因此如果在不同的硬件上运行,建议您尝试使用此选项。
对于本文描述的实验,我们使用独立启动的同一模拟系统的多个实例。这是真实集合模拟的代理,当然,成员模拟和不规则通信之间可能存在细微差异,但仍有以类似方式重叠的范围。
MPS
随着 GPU 的规模和功能不断增加,单个应用程序的执行可能无法充分利用 GPU 提供的所有资源。 NVIDIA 多进程服务( MPS )是一种使多个 CPU 进程提交的计算内核能够在同一 GPU 上同时执行的工具。这种重叠可能实现更彻底的资源使用和更好的总体吞吐量。
使用 MPS 还可以通过更有效地重叠硬件资源利用率和更好地利用基于 CPU 的并行性,在多个 GPU 之间实现应用程序的强大扩展,如本文后面所述。
2017 年, NVIDIA Volta 架构与 NVIDIA Volta MPS 一起发布,具有增强的功能,包括每个 GPU 最多增加 48 个 MPS 客户端:这在所有后续 V100 、 Quadro 和 GeForce GPU 上都受支持。
MIG
与 MPS 一样, NVIDIA 多实例 GPU ( MIG )促进了每个 GPU 的共享,但对资源进行了严格的分区。它非常适合在多个不同的用户之间共享 GPU ,因为每个 MIG 实例都有一组有保证的资源,并且是完全隔离的。
GPU 在选定的 NVIDIA 安培体系结构 MIG 上可用,包括 A100 ,每个 GPU 最多支持七个 MIG 实例。 MIG 可以与 MPS 组合,其中多个 MPS 客户端可以在每个 MIG 实例上同时运行,每个物理 GPU 最多支持 48 个 MPS 客户端。
虽然可以跨多个 MIG 实例运行单个应用程序实例,例如使用 MPI , MIG 的目标并不是为该用例提供任何性能改进。 MIG 的主要目的是在每个 GPU 上促进多个不同的应用程序实例。有关更多信息,请参阅通过多实例 GPU 充分利用 NVIDIA A100 GPU 。
绩效结果
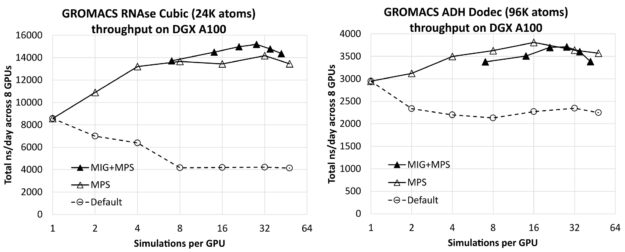
图 1 显示了 RNAse (左)和 ADH (右)在 8- GPU DGX A100 服务器上同时运行的所有模拟中,每个 GPU 的模拟次数对总综合吞吐量(以 ns /天为单位,越高越好)的影响。每种情况下最左边的结果(每个 GPU 进行一次模拟)仅比相应的单独模拟结果(前面给出)乘以 8 ( DGX A100 服务器上 GPU 的数量)低几个百分点。这表明您可以在服务器上有效地运行多个模拟,而不会产生明显的干扰。
您可以看到,通过增加每个 GPU 的模拟次数,可以获得实质性的改进,对于相对较小的 RNAse 病例,模拟次数最多为 1 . 8X ,对于较大的 ADH 病例,模拟次数最多为 1 . 3X 。
对于每个测试用例,我们使用每个 GPU 最多七个 MIG 分区,包括没有 MIG 和有 MIG 的结果。在这种情况下,MPS用于每个 GPU 运行多个进程,每个物理 GPU 最多运行48个MPS客户端,其中 MIG 情况下的最大客户端总数为42:7个 MIG 分区中的每个分区有6个MPS客户端。作为参考,我们还包括了没有MPS或 MIG 的默认结果,对于这些结果,我们认为按照 GPU 运行多个模拟没有任何好处。
对于每个测试用例,最左边的 MIG+MPS 结果是每个 GPU 进行 7 次模拟:每个 MIG 客户端进行一次模拟(即,没有 MPS)。我们发现这些“纯 MIG”性能结果与相应的“纯 MPS”结果相比没有优势。对于 RNAse,纯 MIG 类似于纯 MPS,而对于 ADH 则低于纯 MPS。然而,将 MIG 与 MPS 相结合会导致 RNAse 的最佳整体结果,比最佳纯 MPS 结果高约 7%。它导致性能与 ADH 的纯 MPS 相当,但略低于纯 MPS。
对于 RNAse ,性能最好的配置是每个 MIG 四个 MPS 客户端,即每个 GPU 总共 28 个模拟。对于 ADH ,最好的配置是使用纯 MPS ,每 GPU 进行 16 次模拟,而不使用 MIG 。
当 MIG 处于活动状态时,它强制将每个模拟隔离到 GPU 硬件的特定分区,这可能是有利的,具体取决于测试用例关键路径的特定数据访问模式。另一方面,在没有 MIG 的情况下,每个仿真都可以以更动态的方式访问 GPU 上的资源,这也是有利的。
MIG 的好处可能取决于测试用例,正如我们前面讨论的那样。令人欣慰的是,MPS在有 MIG 和没有 MIG 的情况下都是有效的,特别是在有些用例中 MIG 出于其他原因是可取的,例如在用户之间共享 GPU 。
GROMACS 是异构的,并且具有灵活性,在这方面,计算可以卸载到 GPU ,其中 CPU 可以同时使用。我们的运行使用了力计算的默认选项,将非粘结力和 PME 力计算映射到 GPU ,同时使用 CPU 进行粘结力计算。这种配置通常会导致资源的良好重叠使用。
我们尝试将粘结力计算卸载到 GPU (使用 -bonding GPU 选项),性能类似,但在所有情况下都略低。如前所述,我们使用 GPU – 驻留步骤。我们尝试将更新和约束映射到 CPU ,我们还观察到了根据 GPU 运行多个模拟的好处。
对于较大的 ADH 情况,可实现的吞吐量大大低于将更新和约束卸载到 GPU 时的吞吐量。然而,对于较小的 RNAse 情况,尽管在每 GPU 运行一次(或几次)模拟时吞吐量较低,但在每 GPU 运行八次或更多模拟时,无论是否卸载此部分,我们都看到了类似的吞吐量。行为可能因测试用例和硬件而异,因此最好使用所有可用的运行时组合进行实验。
我们还在不同的硬件上重复了核糖核酸酶的MPS实验:NVIDIA A40和V100-SXM2 GPU ,我们还发现,每个 GPU 运行多个模拟可以提高吞吐量,尽管程度低于A100。鉴于这些 GPU 的规格相对较低,这并不奇怪。A40为1.5倍,V100-SXM2为1.4倍,观察到的吞吐量改善明显低于A100的1.8倍,但仍然值得。
这些结果表明,通过在 MPS 中按 GPU 运行多个进程,并将 MIG 与 MPS 相结合,可以实现大的吞吐量改进。最佳配置(包括 GROMACS 中的计算卸载选项)取决于具体情况,我们再次建议进行实验。以下各节描述了这些模拟是如何编排的。
运行配置详细信息
在本节中,我们将提供用于生成结果的脚本,并描述其中包含的命令,作为您自己工作流的参考或起点。
纯议员选举
以下脚本使用 MPS 向 8- GPU DGX A100 服务器启动多个模拟。
1 #!/bin/bash
2 # Demonstrator script to run multiple simulations per GPU with MPS on DGX-A100
3 #
4 # Alan Gray, NVIDIA
5
6 # Location of GROMACS binary
7 GMX=/lustre/fsw/devtech/hpc-devtech/alang/gromacs-binaries/v2021.2_tmpi_cuda11.2/bin/gmx
8 # Location of input file
9 INPUT=/lustre/fsw/devtech/hpc-devtech/alang/Gromacs_input/rnase.tpr
10
11 NGPU=8 # Number of GPUs in server
12 NCORE=128 # Number of CPU cores in server
13
14 NSIMPERGPU=16 # Number of simulations to run per GPU (with MPS)
15
16 # Number of threads per simulation
17 NTHREAD=$(($NCORE/($NGPU*$NSIMPERGPU)))
18 if [ $NTHREAD -eq 0 ]
19 then
20 NTHREAD=1
21 fi
22 export OMP_NUM_THREADS=$NTHREAD
23
24 # Start MPS daemon
25 nvidia-cuda-mps-control -d
26
27 # Loop over number of GPUs in server
28 for (( i=0; i<$NGPU; i++ ));
29 do
30 # Set a CPU NUMA specific to GPU in use with best affinity (specific to DGX-A100)
31 case $i in
32 0)NUMA=3;;
33 1)NUMA=2;;
34 2)NUMA=1;;
35 3)NUMA=0;;
36 4)NUMA=7;;
37 5)NUMA=6;;
38 6)NUMA=5;;
39 7)NUMA=4;;
40 esac
41
42 # Loop over number of simulations per GPU
43 for (( j=0; j<$NSIMPERGPU; j++ ));
44 do
45 # Create a unique identifier for this simulation to use as a working directory
46 id=gpu${i}_sim${j}
47 rm -rf $id
48 mkdir -p $id
49 cd $id
50
51 ln -s $INPUT topol.tpr
52
53 # Launch GROMACS in the background on the desired resources
54 echo "Launching simulation $j on GPU $i with $NTHREAD CPU thread(s) on NUMA region $NUMA"
55 CUDA_VISIBLE_DEVICES=$i numactl --cpunodebind=$NUMA $GMX mdrun \
56 -update gpu -nsteps 10000000 -maxh 0.2 -resethway -nstlist 100 \
57 > mdrun.log 2>&1 &
58 cd ..
59 done
60 done
61 echo "Waiting for simulations to complete..."
62 wait
- 第 7 行和第 9 行分别指定 GROMACS 二进制文件和测试用例输入文件的位置。
- 第 11-12 行指定服务器的固定硬件详细信息,该服务器有 8 个 GPU 和 128 个 CPU 内核。
- 第 14 行指定了每个 GPU 的模拟次数,可以改变模拟次数以评估性能,正如生成上述结果所做的那样。
- 第 17-21 行计算每个模拟应分配多少 CPU 个线程。
- 第 25 行启动 MPS 守护进程,使从单独模拟启动的内核能够在同一 GPU 上同时执行。
- 第 28-40 行在服务器中的 GPU 上循环,并为每个特定 GPU 分配一组适当的 CPU 内核(“NUMA区域”)。此映射特定于DGX A100拓扑,该拓扑具有两个AMD CPU s,每个AMD CPU 具有四个NUMA区域。我们安排特定的编号以获得最佳亲和力。有关更多信息,请参见DGX A100 用户指南中的第1.3节。
- 第 43-49 行循环每个 GPU 的模拟次数,并创建模拟特有的工作目录。
- 第 51 行在此唯一工作目录中创建指向输入文件的链接。
- 第 55-57 行启动每个模拟,使用
CUDA_VISIBLE_DEVICES环境变量将其限制为所需的 GPU ,使用 numactl 实用程序将其限制为所需的 CPU NUMA 区域。可以使用< code > apt install numactl </ code >提前安装该实用程序。
-update GPU 选项与默认的 GPU 强制卸载行为相结合,对性能至关重要(见上文),而-nsteps 10000000 -maxh 0.2 -resethway组合的结果是运行每个模拟 12 分钟( 0 . 2 小时),其中内部计时器会在中途重置,以消除任何初始化开销。-nstlist 100指定 GROMACS 应每隔 100 步重新生成内部邻居列表,其中这是一个可调参数,影响性能但不影响正确性。)
结合 MIG 和 MPS 运行
以下脚本是前一个脚本的版本,扩展为支持每个 GPU 的多个 MIG 实例,其中每个 MIG 实例可以使用 MPS 启动多个仿真。
1 #!/bin/bash
2 # Demonstrator script to run multiple simulations per GPU with MIG+MPS on DGX-A100
3 #
4 # Alan Gray, NVIDIA
5
6 # Location of GROMACS binary
7 GMX=/lustre/fsw/devtech/hpc-devtech/alang/gromacs-binaries/v2021.2_tmpi_cuda11.2/bin/gmx
8 # Location of input file
9 INPUT=/lustre/fsw/devtech/hpc-devtech/alang/Gromacs_input/adhd.tpr
10
11 NGPU=8 # Number of GPUs in server
12 NCORE=128 # Number of CPU cores in server
13
14 NMIGPERGPU=7 # Number of MIGs per GPU
15 NSIMPERMIG=3 # Number of simulations to run per MIG (with MPS)
16
17 # Number of threads per simulation
18 NTHREAD=$(($NCORE/($NGPU*$NMIGPERGPU*$NSIMPERMIG)))
19 if [ $NTHREAD -eq 0 ]
20 then
21 NTHREAD=1
22 fi
23 export OMP_NUM_THREADS=$NTHREAD
24
25 # Loop over number of GPUs in server
26 for (( i=0; i<$NGPU; i++ ));
27 do
28 # Set a CPU NUMA specific to GPU in use with best affinity (specific to DGX-A100)
29 case $i in
30 0)NUMA=3;;
31 1)NUMA=2;;
32 2)NUMA=1;;
33 3)NUMA=0;;
34 4)NUMA=7;;
35 5)NUMA=6;;
36 6)NUMA=5;;
37 7)NUMA=4;;
38 esac
39
40 # Discover list of MIGs that exist on this GPU
41 MIGS=`nvidia-smi -L | grep -A $(($NMIGPERGPU+1)) "GPU $i" | grep MIG | awk '{ print $6 }' | sed 's/)//g'`
42 MIGARRAY=($MIGS)
43
44 # Loop over number of MIGs per GPU
45 for (( j=0; j<$NMIGPERGPU; j++ ));
46 do
47
48 MIGID=${MIGARRAY[j]}
49 # Start MPS daemon unique to MIG
50 export CUDA_MPS_PIPE_DIRECTORY=/tmp/$MIGID
51 mkdir -p $CUDA_MPS_PIPE_DIRECTORY
52 CUDA_VISIBLE_DEVICES=$MIGID nvidia-cuda-mps-control -d
53
54 # Loop over number of simulations per MIG
55 for (( k=0; k<$NSIMPERMIG; k++ ));
56 do
57
58 # Create a unique identifier for this simulation to use as a working directory
59 id=gpu${i}_mig${j}_sim${k}
60 rm -rf $id
61 mkdir -p $id
62 cd $id
63
64 ln -s $INPUT topol.tpr
65
66 # Launch GROMACS in the background on the desired resources
67 echo "Launching simulation $k on MIG $j, GPU $i with $NTHREAD CPU thread(s) on NUMA region $NUMA"
68 CUDA_VISIBLE_DEVICES=$MIGID numactl --cpunodebind=$NUMA $GMX mdrun \
69 -update gpu -nsteps 10000000 -maxh 0.2 -resethway -nstlist 100 \
70 > mdrun.log 2>&1 &
71 cd ..
72 done
73 done
74 done
75 echo "Waiting for simulations to complete..."
76 wait
与纯 MPS 脚本的主要区别以粗体突出显示:
- 第 14 行指定每个 GPU 的 MIG 实例数,设置为最多 7 个。使用以下命令提前在八个 GPU 中的每一个上创建实例:
for gpu in 0 1 2 3 4 5 6 7
do
nvidia-smi mig -i $gpu --create-gpu-instance \
1g.10gb,1g.10gb,1g.10gb,1g.10gb,1g.10gb,1g.10gb,1g.10gb \
--default-compute-instance
done
- 第 15 行指定每个 MIG 实例要运行的模拟数量。
- 第 18 行调整 CPU 核心分配计算,以正确地考虑每个 GPU 的总模拟次数。
- 第 41-42 行位于循环(物理) GPU s 内,列出与 GPU 关联的 MIG s 唯一的 ID ,并创建包含这七个 ID 的可索引数组。
- 第 45 行向循环嵌套添加一个新的中间级别,对应于每个 GPU 的多个 MIG s 。
- 第 48-52 行根据 MIG 启动一个单独的 MPS 守护程序,这是结合 MIG 和 MPS 的要求。
- 第 55 行在每个 MIG 的模拟次数上循环。它继续像以前一样启动每个模拟,但现在每个模拟都被限制为使用唯一 MIG ID 的特定 MIG 实例。
多流程的其他优势
到目前为止,我们已经向您展示了如何按照 GPU 运行多个进程可以为 GROMACS 带来巨大的好处,并且我们提供了具体的示例来演示。类似的技术可以在更广泛的用例和场景中提供好处,包括 GROMACS 和其他应用程序。在本节中,我们将简要讨论其中的一些。
GROMACS 多模拟框架
在这篇文章中,我们使用 shell 脚本中的循环并行启动了多个模拟。内置的 GROMACS 多仿真框架提供了一种替代机制,其中通过-multidir选择mdrun启动 GROMACS 时,将多个 MPI 任务映射到多个仿真。以类似的方式,最大化吞吐量的好处也可以应用于该机制。
为了评估这一点,我们没有直接使用 MPI 启动 GROMACS ,而是通过一个包装器脚本启动 GROMACS ,该脚本可以为每个 MPI 级别设置适当的环境变量和numactl选项。可以使用环境变量发现秩,例如 OpenMPI 的OMPI_COMM_WORLD_LOCAL_RANK。另一种方法是以类似的方式使用 MPI 启动器的绑定功能。
我们的实验表明, MPS 的行为与前面描述的一样。然而,相比之下,我们并没有看到 MPS 和 MIG 结合的任何额外好处,这需要进一步的研究。
多模拟框架还支持在集合内的模拟之间进行不频繁的副本交换,使用-replex选择mdrun或AWH 多步行机方法在一个模拟中应用多个独立的偏置电位。对于这些工作流,性能行为取决于用例的特定细节,因此我们建议进行实验。有关-replex选项的更多信息,请参阅副本交换模拟简介教程。
用于多重 GPU 强扩展的 MPS
这篇文章探讨了 MPS (和 MIG )的好处,以提高并行运行的许多独立模拟的吞吐量。另一个共同目标是通过并行使用多个 GPU 来最小化单个模拟的求解时间。
通常,单独的 CPU 任务(如 MPI 任务)用于控制每个 GPU ,并执行未卸载到 GPU 的任何计算工作负载。在某些情况下,每个 GPU 运行多个 CPU 任务是有益的,因为这可以为重叠的 CPU 计算 GPU 计算和通信提供额外的机会。按照 GPU 运行多个进程也有助于重新平衡并行运行中固有的负载不平衡。有关更多信息,请参阅本节后面讨论的 GROMACS 示例。
此方法还可以增加 CPU 上基于任务的并行程度,并增强应用程序中任何 CPU 驻留的并行工作负载的性能。当 MPS 处于活动状态时,与多个任务关联的多个内核可以在每个 GPU 上并行执行。最好是进行实验,以发现它是否能对特定情况有益,并找到最佳配置。
格罗马克的例子
下面是一个具体的例子。在以前的职位中,我们将重点放在每个 GROMACS 模拟并行运行四个 GPU 的情况下,以最小化求解时间。我们展示了这四个 GPU 如何有效地平衡三个 PP 任务和一个 PME 任务。
但是,如果您试图将该配置调整为两个 GPU ,最自然的方法是分配一个 PP GPU 和一个 PME GPU 。这不会产生良好的性能,因为 PP GPU 的工作量要大得多。
最好将四 GPU 配置映射到两 GPU 配置,并激活 MPS 以启用内核重叠。 GPU 中的一个重叠两个 PP 任务,而另一个重叠两个 PP 和一个 PME 。这将导致更好的负载平衡和更快的解决时间。在GROMACS 中分子动力学模拟的非均匀并行化和加速论文中,该技术用于生成图 12 的两个 GPU 强缩放结果。同样,我们建议对任何特定情况进行实验。
与计算重叠的 I / O
这篇文章展示了按照 GPU 运行多个进程如何通过重叠提供好处。好处不仅限于计算和通信。此解决方案也适用于花费大量 I / O 时间的情况。在这些情况下,一个实例的 I / O 组件可以与另一个实例的计算组件重叠,以提高总体吞吐量。 MPS 使内核能够与其他计算内核、通信或文件 I / O 并行执行。
如果您有问题或建议,请在下面进行评论。