GROMACS, a simulation package for biomolecular systems, is one of the most highly used scientific software applications worldwide, and a key tool in understanding important biological processes including those underlying the current COVID-19 pandemic.
In a previous post, we showcased recent optimizations, performed in collaboration with the core development team, that enable GROMACS to operate much faster than previously possible on modern multi-GPU servers. These optimizations involve offloading both computation and communication to the GPU, where the latter is of particular benefit to those relatively large cases that can effectively use multiple GPUs in parallel for a single simulation. For more information about the parallelization and inner workings of the latest heterogeneous software engine, see Heterogeneous parallelization and acceleration of molecular dynamics simulations in GROMACS.
Another increasingly common workflow involves running many independent GROMACS simulations, where each simulation can be relatively small. NVIDIA GPUs continue to increase in size and capability, but it can often be the case that isolated single-trajectory simulations of relatively small simulation systems can’t fully use all the available computational resources on each GPU.
But multitrajectory workflows can involve anywhere from tens to thousands of loosely coupled molecular dynamics simulations. In such workloads, the aim most often is not to minimize time-to-solution for a single simulation but instead maximize throughput for the entire ensemble.
Running multiple simulations per GPU in parallel can substantially increase overall throughput, as has been previously shown for GROMACS in Heterogeneous parallelization and acceleration of molecular dynamics simulations in GROMACS (Figure 11). The NVIDIA Multi-Process Server (MPS) and Multi-Instance GPU (MIG) features have been created to facilitate such workflows, further enhancing efficiency by enabling each GPU to be used for multiple tasks simultaneously.
In this post, we demonstrate the benefits of running multiple simulations per GPU for GROMACS. We show how to take advantage of MPS, including in combination with MIG, to run multiple simulations on each GPU in parallel, achieving up to 1.8X overall improvement in throughput.
Background information
Hardware
For the results given later in this post, we ran on a DGX A100 server, part of the NVIDIA-internal Selene supercomputer, which features eight A100-SXM4-80GB GPUs and two AMD EPYC 7742 (Rome) 64-core CPU sockets.
GROMACS test cases
To generate the results given in the following section, we use the 24K atom RNAse cubic and the 96K atom ADH dodec test cases, both of which use PME for long-range electrostatics. For more information and input files, see Supplementary Information for Heterogeneous Parallelization and Acceleration of Molecular Dynamics Simulations in GROMACS.
We use GROMACS version 2021.2 (with CUDA Version 11.2, Driver Version 470.57.02). For a single isolated instance (on a single A100-SXM4-80GB GPU), using the same GROMACS options as described later, we achieved 1,083 ns/day for RNAse and 378 ns/day for ADH.
The -update gpu option is crucial to performance here. This triggers the “GPU-resident step,” where the update and constraints part of each timestep is offloaded to the GPU along with the default “GPU force offload” behavior. Without this, the performance is around a factor of 2 lower for each case, on this hardware. This is sensitive to the balance of CPU and GPU capabilities available, so we recommend that you experiment with this option if running on different hardware.
For the experiments described in this post, we use multiple instances of the same simulation system launched independently. This is a proxy for real ensemble simulations, where of course there can be minor differences between member simulation as well as irregular communication, but still scope to overlap in a similar fashion.
MPS
As GPUs continue to increase in size and capability, a single application execution may not be able to fully use all the resources available per GPU. NVIDIA Multi-Process Service (MPS) is a facility that enables compute kernels submitted from multiple CPU processes to execute simultaneously on the same GPU. Such overlapping can potentially enable more thorough resource use and better overall throughput.
Using MPS can also benefit the strong scaling of applications across multiple GPUs, through more efficient overlapping of hardware resource utilization and better exploitation of CPU-based parallelism, as described later in this post.
In 2017, the NVIDIA Volta architecture was released accompanied by NVIDIA Volta MPS with enhanced capabilities including an increased maximum of 48 MPS clients per GPU: this is supported on all subsequent V100, Quadro, and GeForce GPUs.
MIG
Like MPS, NVIDIA Multi-Instance GPU (MIG) facilitates the sharing of each GPU, but with strict partitioning of resources. It is ideal for sharing a GPU across multiple different users, as each MIG instance has a guaranteed set of resources and is fully isolated.
MIG is available on selected NVIDIA Ampere Architecture GPUs, including A100, which supports a maximum of seven MIG instances per GPU. MIG can be combined with MPS, where multiple MPS clients can run simultaneously on each MIG instance, up to a maximum of 48 total MPS clients per physical GPU.
While it is possible to run a single application instance across multiple MIG instances, such as with MPI, MIG does not aim to offer any performance improvements for this use case. The primary aim of MIG is to facilitate multiple distinct application instances on each GPU. For more information, see Getting the Most Out of the NVIDIA A100 GPU with Multi-Instance GPU.
Performance results
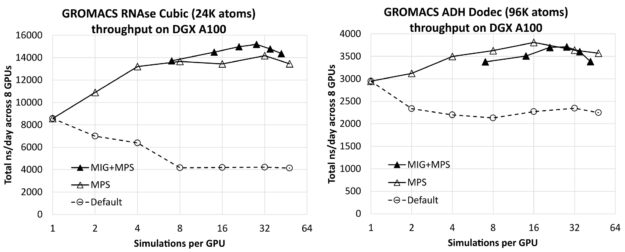
Figure 1 shows the effect of the number of simulations per GPU on the total combined throughput (in ns/day, higher is better) of all simulations running simultaneously across the 8-GPU DGX A100 server, for RNAse (left) and ADH (right). The leftmost result for each case (one simulation per GPU) is only a few percent lower than the corresponding isolated single-simulation result (given earlier) multiplied by 8 (the number of GPUs on the DGX A100 server). This shows that you can effectively run multiple simulations across the server without significant interference.
You can see that substantial improvement is available through increasing the number of simulations per GPU, up to a factor of 1.8X for the relatively small RNAse case and a factor of 1.3X for the larger ADH case.
For each test case, we included results without and with MIG, using the maximum of seven MIG partitions per GPU. In this case, MPS was used to run multiple processes per GPU up to the maximum of 48 MPS clients per physical GPU, where the maximum total number of clients in the MIG case was 42: Six MPS clients for each of the seven MIG partitions. For reference, we also included default results without either MPS or MIG, for which we saw no benefit from running multiple simulations per GPU.
For each test case, the leftmost MIG+MPS results are at seven simulations per GPU: One simulation per MIG client (that is, without MPS). We find that these “pure-MIG” performance results have no advantage over the corresponding “pure-MPS” results. Pure-MIG is similar to pure-MPS for RNAse, and lower than pure-MPS for ADH. However, combining MIG with MPS leads to the best overall result for RNAse, by around 7% over the best pure-MPS result. It leads to performance comparable with, but slightly lower than, pure-MPS for ADH.
For RNAse, the best performing configuration is four MPS clients per MIG, that is, a total of 28 simulations per GPU. For ADH, the best performing configuration is 16 simulations per GPU using pure-MPS, without MIG.
When MIG is active, it enforces each simulation to be isolated to a specific partition of the GPU hardware, which can be advantageous depending on the specific data access patterns for the critical path of the test case. On the other hand, without MIG, each simulation can access resources across the GPU in a more dynamic manner, which can also be advantageous.
The benefit of MIG can depend on the test case, as we discussed earlier. It is reassuring to see that MPS is effective with and without MIG, especially as there are some use cases where MIG is desirable for other reasons, for example sharing a GPU between users.
GROMACS is heterogeneous and has flexibility in terms of which calculations are offloaded to the GPU, where the CPU can be used concurrently. Our runs used the default options for force calculations, which map nonbonded and PME force calculations to GPU, while using the CPU for bonded force calculations. This configuration often results in good overlapping usage of resources.
We tried instead offloading the bonded force calculations to GPU (with the -bonded gpu option), and performance was similar but slightly lower for all cases. As noted earlier, we used the GPU-resident step. We tried instead mapping update and constraints to the CPU, where we also observed benefits from running multiple simulations per GPU.
For the larger ADH case, the achievable throughput is substantially lower than when update and constraints are offloaded to GPU. However, for the smaller RNAse case, although the throughput is lower when running a single (or a few) simulations per GPU, at eight or more simulations per GPU, we saw similar throughput whether this part is offloaded. The behavior may vary across test cases and hardware, so it is always a good idea to experiment with all available runtime combinations.
We also repeated the MPS experiments for RNAse on different hardware: the NVIDIA A40 and V100-SXM2 GPUs, where we also found running multiple simulations per GPU to offer throughput improvements, albeit at lesser extent than A100. This is not surprising given the relatively lower specifications of these GPUs. At 1.5X for A40 and 1.4X for V100-SXM2, the observed improvements in throughput are significantly lower than the 1.8X observed for A100, but still worthwhile.
These results show that large throughput improvements can be realized through running multiple processes per GPU with MPS and combining MIG with MPS is effective. The best configuration, including computational offload options within GROMACS, is dependent on the case, and we again recommend experimentation. The following sections describe how these simulations were orchestrated.
Run configuration details
In this section, we provide the scripts used to generate the results, and describe the commands contained within, as a reference or starting point for your own workflow.
Pure MPS runs
The following script launches multiple simulations with MPS to the 8-GPU DGX A100 server.
1 #!/bin/bash
2 # Demonstrator script to run multiple simulations per GPU with MPS on DGX-A100
3 #
4 # Alan Gray, NVIDIA
5
6 # Location of GROMACS binary
7 GMX=/lustre/fsw/devtech/hpc-devtech/alang/gromacs-binaries/v2021.2_tmpi_cuda11.2/bin/gmx
8 # Location of input file
9 INPUT=/lustre/fsw/devtech/hpc-devtech/alang/Gromacs_input/rnase.tpr
10
11 NGPU=8 # Number of GPUs in server
12 NCORE=128 # Number of CPU cores in server
13
14 NSIMPERGPU=16 # Number of simulations to run per GPU (with MPS)
15
16 # Number of threads per simulation
17 NTHREAD=$(($NCORE/($NGPU*$NSIMPERGPU)))
18 if [ $NTHREAD -eq 0 ]
19 then
20 NTHREAD=1
21 fi
22 export OMP_NUM_THREADS=$NTHREAD
23
24 # Start MPS daemon
25 nvidia-cuda-mps-control -d
26
27 # Loop over number of GPUs in server
28 for (( i=0; i<$NGPU; i++ ));
29 do
30 # Set a CPU NUMA specific to GPU in use with best affinity (specific to DGX-A100)
31 case $i in
32 0)NUMA=3;;
33 1)NUMA=2;;
34 2)NUMA=1;;
35 3)NUMA=0;;
36 4)NUMA=7;;
37 5)NUMA=6;;
38 6)NUMA=5;;
39 7)NUMA=4;;
40 esac
41
42 # Loop over number of simulations per GPU
43 for (( j=0; j<$NSIMPERGPU; j++ ));
44 do
45 # Create a unique identifier for this simulation to use as a working directory
46 id=gpu${i}_sim${j}
47 rm -rf $id
48 mkdir -p $id
49 cd $id
50
51 ln -s $INPUT topol.tpr
52
53 # Launch GROMACS in the background on the desired resources
54 echo "Launching simulation $j on GPU $i with $NTHREAD CPU thread(s) on NUMA region $NUMA"
55 CUDA_VISIBLE_DEVICES=$i numactl --cpunodebind=$NUMA $GMX mdrun \
56 -update gpu -nsteps 10000000 -maxh 0.2 -resethway -nstlist 100 \
57 > mdrun.log 2>&1 &
58 cd ..
59 done
60 done
61 echo "Waiting for simulations to complete..."
62 wait
- Lines 7 and 9 specify the location of the GROMACS binary, and the test case input file, respectively.
- Lines 11-12 specify fixed hardware details of the server, which has 8 GPUs and 128 CPU cores.
- Line 14 specifies the number of simulations per GPU, which can be varied to assess performance, as was done to generate the above results.
- Lines 17-21 works out how many CPU threads should be assigned to each simulation.
- Line 25 starts the MPS daemon, which enables kernels launched from separate simulations to execute simultaneously on the same GPU.
- Lines 28-40 loop over the GPUs in the server and assign an appropriate set of CPU cores (a “NUMA region”), appropriate to each specific GPU. This mapping is specific to the DGX A100 topology, which has two AMD CPUs, each with four NUMA regions. We arrange the specific numbering for optimal affinity. For more information, see Section 1.3 in the DGX A100 User Guide.
- Lines 43-49 loop over the number of simulations per GPU and create a working directory unique to a simulation.
- Line 51 creates a link to the input file within this unique working directory.
- Lines 55-57 launch each simulation, which is restricted to the desired GPU using the
CUDA_VISIBLE_DEVICESenvironment variable, and the desired CPU NUMA region using the numactl utility. The utility can be installed in advance with <code>apt install numactl</code>.
The -update gpu option, in combination with the default GPU force offload behavior, is crucial for performance (see earlier), and the -nsteps 10000000 -maxh 0.2 -resethway combination has the result of running each simulation for 12 minutes (0.2 hours), where the internal timers are reset halfway to remove any initialization overhead. The -nstlist 100 specifies that GROMACS should regenerate the internal neighbor list every 100 steps, where this is a tunable parameter that affects performance but not correctness.)
Runs combining MIG and MPS
The following script is a version of the previous one, extended to support multiple MIG instances per GPU, where multiple simulations can be launched per MIG instance with MPS.
1 #!/bin/bash
2 # Demonstrator script to run multiple simulations per GPU with MIG+MPS on DGX-A100
3 #
4 # Alan Gray, NVIDIA
5
6 # Location of GROMACS binary
7 GMX=/lustre/fsw/devtech/hpc-devtech/alang/gromacs-binaries/v2021.2_tmpi_cuda11.2/bin/gmx
8 # Location of input file
9 INPUT=/lustre/fsw/devtech/hpc-devtech/alang/Gromacs_input/adhd.tpr
10
11 NGPU=8 # Number of GPUs in server
12 NCORE=128 # Number of CPU cores in server
13
14 NMIGPERGPU=7 # Number of MIGs per GPU
15 NSIMPERMIG=3 # Number of simulations to run per MIG (with MPS)
16
17 # Number of threads per simulation
18 NTHREAD=$(($NCORE/($NGPU*$NMIGPERGPU*$NSIMPERMIG)))
19 if [ $NTHREAD -eq 0 ]
20 then
21 NTHREAD=1
22 fi
23 export OMP_NUM_THREADS=$NTHREAD
24
25 # Loop over number of GPUs in server
26 for (( i=0; i<$NGPU; i++ ));
27 do
28 # Set a CPU NUMA specific to GPU in use with best affinity (specific to DGX-A100)
29 case $i in
30 0)NUMA=3;;
31 1)NUMA=2;;
32 2)NUMA=1;;
33 3)NUMA=0;;
34 4)NUMA=7;;
35 5)NUMA=6;;
36 6)NUMA=5;;
37 7)NUMA=4;;
38 esac
39
40 # Discover list of MIGs that exist on this GPU
41 MIGS=`nvidia-smi -L | grep -A $(($NMIGPERGPU+1)) "GPU $i" | grep MIG | awk '{ print $6 }' | sed 's/)//g'`
42 MIGARRAY=($MIGS)
43
44 # Loop over number of MIGs per GPU
45 for (( j=0; j<$NMIGPERGPU; j++ ));
46 do
47
48 MIGID=${MIGARRAY[j]}
49 # Start MPS daemon unique to MIG
50 export CUDA_MPS_PIPE_DIRECTORY=/tmp/$MIGID
51 mkdir -p $CUDA_MPS_PIPE_DIRECTORY
52 CUDA_VISIBLE_DEVICES=$MIGID nvidia-cuda-mps-control -d
53
54 # Loop over number of simulations per MIG
55 for (( k=0; k<$NSIMPERMIG; k++ ));
56 do
57
58 # Create a unique identifier for this simulation to use as a working directory
59 id=gpu${i}_mig${j}_sim${k}
60 rm -rf $id
61 mkdir -p $id
62 cd $id
63
64 ln -s $INPUT topol.tpr
65
66 # Launch GROMACS in the background on the desired resources
67 echo "Launching simulation $k on MIG $j, GPU $i with $NTHREAD CPU thread(s) on NUMA region $NUMA"
68 CUDA_VISIBLE_DEVICES=$MIGID numactl --cpunodebind=$NUMA $GMX mdrun \
69 -update gpu -nsteps 10000000 -maxh 0.2 -resethway -nstlist 100 \
70 > mdrun.log 2>&1 &
71 cd ..
72 done
73 done
74 done
75 echo "Waiting for simulations to complete..."
76 wait
The main differences from the pure-MPS script are highlighted in bold:
- Line 14 specifies the number of MIG instances per GPU, set to the maximum of 7. The instances are created in advance on each of eight GPUs with the following commands:
for gpu in 0 1 2 3 4 5 6 7
do
nvidia-smi mig -i $gpu --create-gpu-instance \
1g.10gb,1g.10gb,1g.10gb,1g.10gb,1g.10gb,1g.10gb,1g.10gb \
--default-compute-instance
done
- Line 15 specifies how many simulations to run per MIG instance.
- Line 18 adjusts the CPU core assignment calculation to properly consider the total number of simulations per GPU.
- Lines 41-42, which is within the loop over (physical) GPUs, lists the IDs unique to the MIGs associated with that GPU, and creates an indexable array containing those seven IDs.
- Line 45 adds a new intermediate level to the loop nest, corresponding to the multiple MIGs per GPU.
- Lines 48-52 starts a separate MPS daemon per MIG, which is a requirement of combining MIG and MPS.
- Line 55 loops over the number of simulations per MIG. It goes on to launch each simulation as before, except now each simulation is restricted to a specific MIG instance using the unique MIG ID.
Other advantages of multiple processes
So far, we’ve shown you how running multiple processes per GPU can offer substantial benefits for GROMACS, and we have provided specific examples to demonstrate. Similar techniques can offer benefits across a wider range of use cases and scenarios, involving GROMACS and other applications. We briefly discuss a few of these in this section.
GROMACS multisimulation framework
For this post, we launched multiple simulations in parallel using loops in shell scripts. The GROMACS in-built multi-simulation framework offers an alternative mechanism, where GROMACS is launched with multiple MPI tasks that are mapped to multiple simulations, through the -multidir option to mdrun. The benefits to maximize throughput can also be applied to this mechanism, in an analogous way.
To assess this, instead of launching GROMACS directly with MPI, we launched through a wrapper script that can set environment variables and numactl options appropriately for each MPI rank. Rank can be discovered with an environment variable, for example OMPI_COMM_WORLD_LOCAL_RANK for OpenMPI. An alternative method is to use binding capabilities of the MPI launcher in a similar fashion.
Our experiments showed that the behavior with MPS is like that described earlier. However, in contrast, we didn’t see any additional benefits by combining MPS and MIG, which requires further investigation.
The multisimulation framework also enables infrequent replica exchange between simulations within the ensemble with the -replex option to mdrun or the AWH multi-walker method to apply multiple independent bias potentials within one simulation. For these workflows, performance behavior is dependent on the specific details of the use case, so we recommend experimentation. For more information about the -replex option, see An introduction to replica exchange simulations.
MPS for multi-GPU strong scaling
This post explored the benefits of MPS (and MIG), to improve throughput of many independent simulations running in parallel. Another common aim is to minimize time to solution for a single simulation, by using multiple GPUs in parallel.
Often, a separate CPU task, such as an MPI task, is used to control each GPU and perform any compute workload that is not offloaded to the GPU. In some cases, it can be beneficial to run multiple CPU tasks per GPU, as this can offer additional opportunities for overlapping CPU computation, GPU computation and communication. Running multiple processes per GPU can also help rebalance any load imbalance inherent in parallel runs. For more information, see the GROMACS example discussed later in this section.
This method can also increase the extent of task-based parallelism on the CPU and enhance performance of any CPU-resident parallel workload in the application. With MPS active, multiple kernels associated with multiple tasks can execute in parallel on each GPU. It is always best to experiment to discover whether it can benefit a specific case and to find the best configuration.
GROMACS example
Here’s a concrete example for GROMACS. In a previous post, we focused on the case where each GROMACS simulation is run across four GPUs in parallel to minimize the time to solution. We showed how these four GPUs can effectively balance three PP tasks plus one PME task.
However, if you try to adapt that configuration to two GPUs, the most natural way is to assign one PP GPU and one PME GPU. This does not result in good performance because the PP GPU has much more work.
It is much better to map the four-GPU configuration to the two GPUs, with MPS active to enable kernel overlapping. One of the GPUs overlaps two PP tasks, whereas the other overlaps two PP and one PME. This results in a significantly better load balance and faster time-to-solution. In the Heterogeneous parallelization and acceleration of molecular dynamics simulations in GROMACS paper, this technique is used to generate the two-GPU strong-scaling results for Figure 12. Again, we recommend experimentation for any specific case.
Overlapping I/O with computation
This post showed how running multiple processes per GPU can offer benefits through overlapping. The benefits are not restricted to computation and communication. This solution can also be relevant for those cases that spend a significant fraction of time in I/O. In those cases, the I/O component for one instance can be overlapped with the compute component for another, to improve overall throughput. MPS enables kernels to execute concurrently with other compute kernels, communication, or file I/O.
If you have questions or suggestions, please comment below.