GROMACS—one of the most widely used HPC applications— has received a major upgrade with the release of GROMACS 2020. The new version includes exciting new performance improvements resulting from a long-term collaboration between NVIDIA and the core GROMACS developers.
As a simulation package for biomolecular systems, GROMACS evolves particles using the Newtonian equations of motion. Forces dictate movement: for example, two positively charged ions repel each other. Calculating forces is the most expensive part of the simulation because all pairs of particles can potentially interact, and simulations involve many particles.
GROMACS provides functionality to account for a wide range of different types of force calculations. For most simulations, the three most important classes (in terms of computational expense) are:
- Non-bonded short-range forces: Particles within a certain cutoff range are considered to interact directly.
- Particle Mesh Ewald (PME) long-range forces: For larger distances, the forces are modeled through a scheme where Fourier transforms are used to perform calculations in Fourier space. This is much cheaper than calculating all interactions directly in real space.
- Bonded short-range forces: Also required due to specific behavior of bonds between particles, for example, the harmonic potential when two covalently bonded atoms are stretched.
In previous GROMACS releases, GPU acceleration was already supported for these force classes (the CUDA Fast Fourier Transform library is used within the PME force calculation). The most recent addition was GPU bonded forces in the 2019 series, developed through a previous collaboration between NVIDIA and the core GROMACS developers.
However, there was still a problem. The force calculations have become so fast on modern GPUs that other parts of the simulation have become very significant in term of computational expense, especially when you want to use multiple GPUs for a single simulation.
This post describes the new performance features available in the 2020 version that address this issue. For many typical simulations, the whole timestep can now run on the GPU, avoiding CPU and PCIe bottlenecks. Inter-GPU communication operations can now operate directly between GPU memory spaces. The results from this work demonstrate large performance improvements.
Single-GPU Porting and Optimization
The following diagram (Figure 1) depicts the key parts of a single timestep in a typical GROMACS simulation, when run on a single GPU and the previous 2019 version (timestep time running left to right):
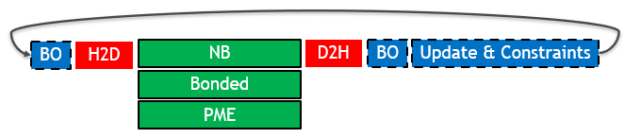
The different box styles indicate the hardware assignment of each part:
- Execution on the GPU (green box with solid border): The three types of force calculation are shown, which can overlap with one another.
- Execution on the CPU (blue box with dashed border): Buffer operations (BO) are internal data layout transformations and force reduction operations. Update & constraints is the final part of the timestep, where atom coordinates are updated and constrained using the previously calculated forces.
- Data transfers (red box with no border): The required PCIe host to device (H2D) and device to host (D2H) data transfers.
GPU acceleration of only the force calculations was an effective strategy on old architectures. Even with acceleration, they were still dominant, such that the other parts of the timestep were not critical in terms of performance. However, on modern architectures (such as Volta), force kernels are now so fast that other overheads have become significant.
The solution in the GROMACS 2020 version is full GPU enablement of all key computational sections. The GPU is used throughout the timestep and repeated PCIe transfers are eliminated, as shown in Figure 2.
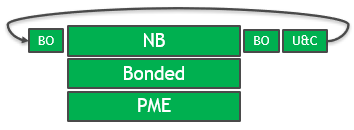
The PME force kernels have also been optimized to allow more efficient assignment of work to CUDA threads and better memory access patterns.
Multi-GPU Optimization
To use multiple GPUs for a single simulation, GROMACS assigns the PME force calculation to a single dedicated GPU. It decomposes the non-bonded and bonded force calculations across the remaining GPUs. A single MPI task will control each GPU: these are called PME tasks and Particle-Particle (PP) tasks, respectively. The following diagram (Figure 3) depicts this for a 4-GPU simulation running the 2019 version:
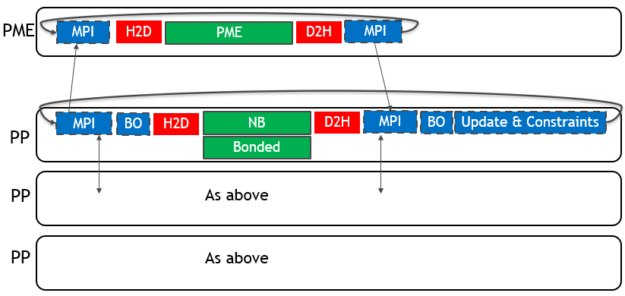
As for the single GPU case, only the force calculations (in green with solid border) are offloaded to the GPU, with CPU (blue with dashed border) and PCIe (red with no border) components limiting the performance.
There are now two types of required communications: those between PP tasks that are of a “halo exchange” nature (where particles at the edge of each domain are transferred to the neighboring domain), and those between PME and PP tasks. Each of these are performed for both coordinate data (near the start of each timestep) and force data (near the end of each timestep).
In this pre-existing code path, the data is routed through the CPU, as shown in the following diagram (Figure 4) for the halo exchange of coordinate data:
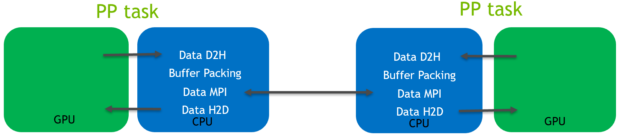
As is shown in the Figure 4 diagram:
- Host to device (H2D) and device to host (D2H) CUDA memory copies were required to transfer data between the GPU and CPU memory spaces. They use the relatively low-bandwidth PCIe bus.
- Buffer packing was performed on the CPU. It was required because data to be transferred is not contiguous.
- MPI calls operating on CPU memory space were used to transfer data between the MPI tasks controlling the separate GPUs.
With GROMACS 2020, these communication operations can now be performed directly between GPU memory spaces and are automatically routed, including using NVLink where available, as shown in Figure 5:
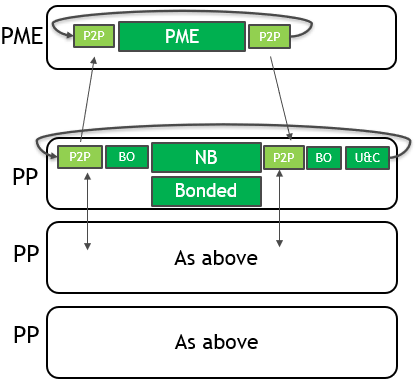
The new strategy is depicted in the following diagram (Figure 6):

CUDA peer-to-peer (P2P) memory copies now transfer data directly between GPU memory spaces. These traverse high-bandwidth, NVLink interconnects where available. Even on systems without NVLink, this new mechanism is substantially faster thanks to the asyncronicity that it offers.
These P2P transfers are facilitated by the fact that the buffer is now directly packed in GPU memory space using a CUDA kernel. This requires information on the array indices involved in the transfer, but the information does not change every timestep. It only changes when the decomposition changes. You infrequently build a “map” of indices on the CPU and transfer to the GPU, without a significant effect on performance.
GROMACS provides an internal MPI library called “thread MPI” that uses CPU threads behind the scenes. Thus, for the main P2P data transfers, you can exploit the fact that multiple MPI tasks are within the same process and use CUDA memory copies operating on the unified virtual address space. However, this means that the optimization is limited to a single compute node (and fallback occurs automatically for unsupported cases). Further work is underway to extend the mechanism to remove this limitation.
Each MPI task must be aware of which remote memory locations to write and read, and must also use remote CUDA events to allow the CUDA streams to synchronize properly across the distinct GPUs. These are exchanged between MPI tasks whenever they change, using MPI send and receive communications.
The earlier diagrams show the halo exchange of coordinate data between PP tasks. The mechanisms for the halo exchange of force data between PP tasks is analogous, where buffer unpacking is required instead of packing. Likewise, the coordinate and force communications between PP and PME tasks are similar, but buffer packing and unpacking is not required as the data is contiguous.
Performance Results
To demonstrate the benefits, we show results from running a multi-GPU simulation for each of three test cases chosen to be representative of the range of system sizes typical to real-world simulations. From smallest to largest, we used the following test-case simulations:
- ADHD (95,561 atoms)
- Cellulose (408,609 atoms)
- STMV (1,066,628 atoms)
We used four NVIDIA V100-SXM2 16GB GPUs (with full NVLink connectivity) hosted by two Intel Xeon E5-2698 v4 (Broadwell) 20-core CPUs, where (as depicted above) we assigned one GPU to PME and decompose PP across the remaining three GPUs.
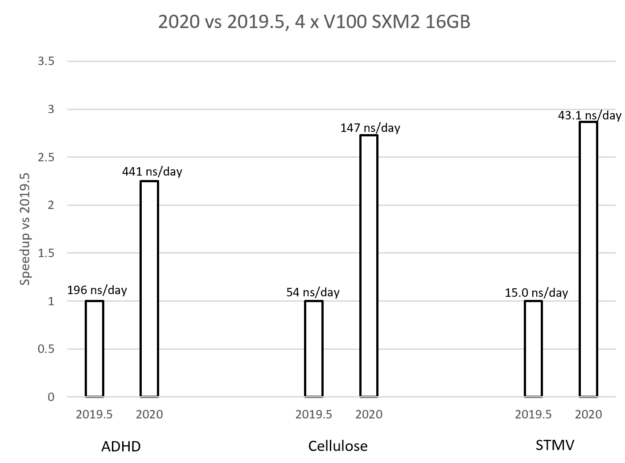
The graph in Figure 7 compares the performance of the new GROMACS 2020 version (with the earlier optimizations enabled) with the previous 2019.5 version, for each of the test cases. The graph shows that the achieved speedup for each case is between 2X and 3X where the speedup increases with the problem size. The labels show the actual performance in each case, given in ns/day, that is, how many nanoseconds can be simulated within a day of running the simulation. These performance benefits are due to the combination of the improvements described in this post.
When running on a single GPU, the multi-GPU communication improvements are of course not relevant so the improvements are less dramatic. However, there can still be significant advantages from the fact that the whole timestep can operate on the GPU with expensive PCIe transfers avoided. When assigning multiple simulations to the same GPU, you may want to use the pre-existing code path depending on the case in use–experiment with the available options.
When running the above performance comparisons, but restricting to one GPU (and one host CPU), we observed the speedup of the 2020 version (with all optimizations enabled) over the previous 2019 version to be in the range of 1.4X – 1.5X across the three test cases.
Enabling Optimizations for Your Simulation
The optimizations in this post are not yet enabled by default. The new code paths have been verified by the standard GROMACS regression tests, but still lack substantial “real-world” testing. We strongly recommend that you carefully verify results against the default path. Any reported issues will be gratefully received by the developer team to help mature the software.
GROMACS should be built using its internal threadMPI library instead of any external MPI library. For more information, see the Multi-GPU Optimization section earlier.
At runtime, the optimizations can be fully enabled by setting the following three environment variables to any non-NULL value in your shell (shown for bash shell here).
For halo exchange communications between PP tasks, use the following command:
export GMX_GPU_DD_COMMS=true
For communications between PME and PP tasks, use the following command:
export GMX_GPU_PME_PP_COMMS=true
To enable the update and constraints part of the timestep for multi-GPU:
export GMX_FORCE_UPDATE_DEFAULT_GPU=true
The combination of these settings triggers all optimizations, including dependencies such as GPU-acceleration of buffer operations.
When running on a single GPU, only GMX_FORCE_UPDATE_DEFAULT_GPU is required (where for single-GPU only, this can alternatively be enabled by adding the -update gpu option to the mdrun command).
In both single and multi GPU cases, it is also necessary to assign the three classes of force calculations to the GPU through the following options to mdrun:
-nb gpu -bonded gpu -pme gpu
On multi-GPU, the -npme 1 option is also required to limit PME to a single GPU.
The new GPU update and constraints code path is only supported in combination with domain decomposition of the PP tasks across multiple GPUs when update groups are used. This means constraining all bonds is not supported, except for small molecules.
To facilitate this, we recommend that you convert bonds with hydrogen bonds to constraints, rather than all bonds. To do this, in the .mdp input file to the GROMACS preprocessor grompp, change the following line:
constraints = all-bonds
The new line should read as follows:
constraints = h-bonds
For more information, see the mdp options section of the User Guide.
Here are the full set of mdrun options that we used when running the 4-GPU performance comparisons:
gmx mdrun -v -nsteps 100000 -resetstep 90000 -noconfout \
-ntmpi 4 -ntomp 10 \
-nb gpu -bonded gpu -pme gpu -npme 1 \
-nstlist 400
The first line instructs GROMACS to run 100,000 steps for this relatively short benchmark test. The timing counters are reset at step 90,000 to avoid the initialization costs being included in the timing, as these are not typically important for long production runs. These specific values have been chosen to allow GROMACS time enough to perform PME tuning automatically, where the size of the PME grid is tuned to give an optimal load balance between PP and PME tasks. Similarly, the -noconfout option instructs GROMACS not to write output files at the end of this short benchmarking run, to avoid artificially high I/O overheads.
The second line specifies that four thread-MPI tasks should be used (one per GPU), with 10 OpenMP threads per thread-MPI task. A total of 40 OpenMP threads are in use to match the number of physical CPU cores in the server.
The third line offloads all force calculations to the GPU, as described earlier.
The final -nstlist 400 option instructs GROMACS to update the neighbor list with a frequency of 400 steps. This option can be adapted without any loss of accuracy when using the Verlet scheme. We found this value to give the best performance through experimentation.
Next Steps
For more information about usage, see the GROMACS 2020 release notes and User Guide. For more information about the developments described in this post, see the GTC 19 presentation Bringing GROMACS up to Speed on Modern Multi-GPU Systems, for which a recording is available.